



INTRODUCCION
En 1974, Schnitzler1 realizó la descripción original del síndrome que lleva su nombre a propósito de un caso de urticaria crónica, lesiones óseas y macroglobulinemia IgM. A partir de entonces se han publicado más de 50 casos de síndrome de Schnitzler, la mayoría de ellos en la literatura francesa2.
Habitualmente se trata de un proceso infradiagnosticado. La presencia de manifestaciones diversas, como la urticaria crónica, la fiebre, los dolores osteo-articulares y las alteraciones hematológicas determinan con frecuencia que el paciente consulte a diversos especialistas, como el dermatólogo, el internista, el reumatólogo o el hematólogo, antes de que se establezca el diagnóstico2.
Presentamos el caso de un paciente en el que se observan la mayoría de las manifestaciones clínicas y serológicas de este raro síndrome.
CASO CLÍNICO
Se trata de una mujer de 76 años de edad, con antecedentes personales de hipertensión arterial, probable alergia a la vitamina B y estenosis aórtica, en tratamiento con amiloride, enalapril, hidroxicina, prednisona, alprazolam, lorazepam, metamizol y para- cetamol, que consultó en el servicio de Dermatología por lesiones cutáneas generalizadas, poco pruriginosas, de aproximadamente dos años de evolución. Cada una de las lesiones, consideradas individualmente, presentaban un curso evanescente, surgiendo a diario y resolviéndose a las pocas horas de forma espontánea sin dejar secuelas. La paciente refería una exacerbación nocturna de la erupción cutánea; además presentaba fiebre nocturna termometrada de hasta 38° C, sin pérdida de peso ni astenia u otros datos de síndrome constitucional asociados, y dolores óseos de intensidad moderada, más importantes a nivel de la columna vertebral y las rodillas. La paciente estaba siendo valorada de forma paralela en el servicio de Hematología por leucotrombocitosis, anemia normocrómica y aumento de la velocidad de sedimentación globular (VSG).
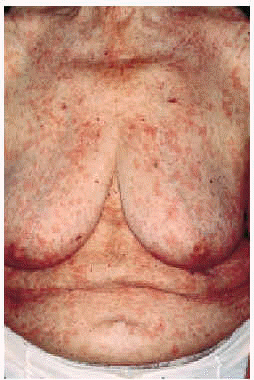
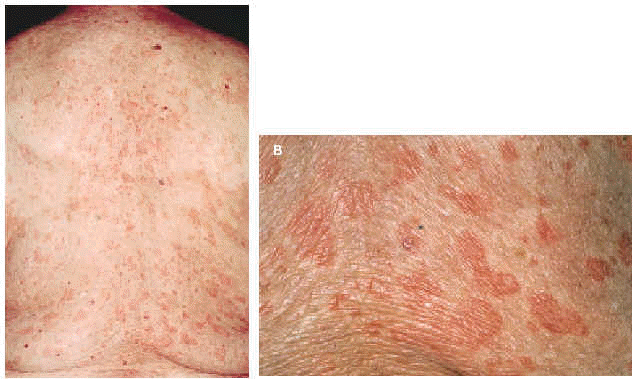
En la exploración física se observaban numerosas lesiones habonosas de tamaño variable entre pocos milímetros y 1 cm de diámetro que afectaban difusamente al tronco y a las extremidades superiores e inferiores (figs. 1 y 2). En los miembros superiores las lesiones se hallaban diseminadas y afectaban incluso las palmas; sin embargo, en los inferiores se hallaban limitadas a la región de los muslos, siendo muy escaso el número de lesiones cutáneas presentes en las piernas. No se palpaban linfadenopatías axilares, inguinales o laterocervicales y tampoco masas ni visceromegalias en la exploración del abdomen.
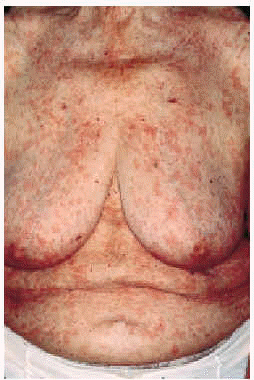
Fig. 1.--Múltiples habones en la parte anterior del tórax.
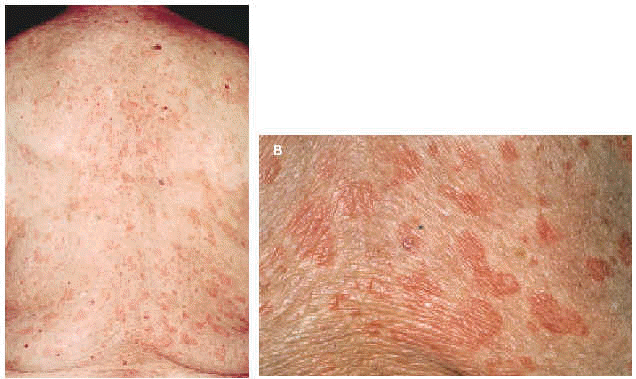
Fig. 2.--A: múltiples habones salpicados por la espalda. B: detalle de los habones.
Se realizó una biopsia de una de las lesiones cutáneas situadas en un antebrazo. El estudio histopatológico reveló la presencia de un infiltrado perivascular de intensidad leve en dermis superficial, compuesto fundamentalmente por neutrófilos, aunque se distinguía algún eosinófilo aislado, sin leucocitoclastia ni daño vascular (fig. 3). Los estudios mediante inmunofluorescencia directa no lograron demostrar depósitos de inmunoglobulinas IgG, IgA, IgM o complemento en las lesiones cutáneas.

Fig. 3.--A: los hallazgos histopatológicos consistían en un infiltrado perivascular e intersticial en la dermis superficial y media, predominantemente constituido por leucocitos, con algún eosinófilo aislado. B: no se observaron hallazgos de vasculitis.
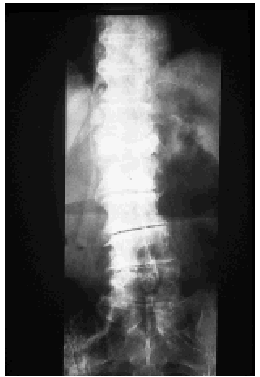
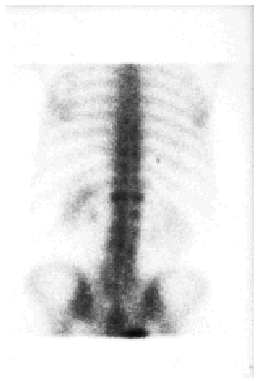
Se llevaron a cabo los siguientes estudios complementarios: el hemograma reveló leucocitosis con neutrofilia (17,7 x 103 leucocitos/mm3 con un 88,5% de neutrófilos), anemia microcítica hipocrómica (hemoglobina de 9,8, con volumen corpuscular medio [VCM] de 71 μm3 y hemoglobina corpuscular media [HCM] de 22,9 pg) sin alteraciones del hierro, la ferritina, la transferrina o la saturación de transferrina, trombocitosis moderada (710 x 103/mm3) y aumento de la VSG (VSG de 90 mm en la primera hora). La bioquímica de sangre y orina fue normal, salvo por un valor discretamente elevado de fosfatasa alcalina sérica. El espectro electroforético del suero reveló un componente monoclonal IgM con cadenas ligeras kappa en la inmunofijación. La cuantificación de inmunoglobulinas detectó una IgM de 1.490 mg/dl, siendo normales las concentraciones de IgG e IgA. No se observaron alteraciones del complemento. Los anticuerpos antinucleares (ANA) y anticuerpos no organoespecíficos (ANOE) fueron negativos durante todo el seguimiento de la paciente, a excepción de una determinación puntual de ANA que resultó positiva a un título de 1/40. La tomografía axial computarizada (TAC) toracoabdominopélvica reveló la presencia de adenopatías bilaterales de pequeño tamaño localizadas en regiones laterocervical y axilar, sin otras anomalías significativas. El estudio de la médula ósea, que se realizó mediante punción esternal, reveló la presencia de una hiperplasia de la serie granulocítica sin alteraciones madurativas y una serie roja normal sin rasgos dishemopoyéticos. No se observó linfocitosis ni plasmocitosis, por lo que se descartó un proceso linfoproliferativo de base. El inmunofenotipo y el análisis citogenético de la médula ósea fueron normales. En la serie ósea se apreciaron acusados signos degenerativos en columna dorsolumbar, así como aumento de la densidad de alguna de las vértebras (fig. 4). En la gammagrafía con tecnecio 99 se observaron dos incrementos focales del trazador en D12 de morfología redondeada (fig. 5).
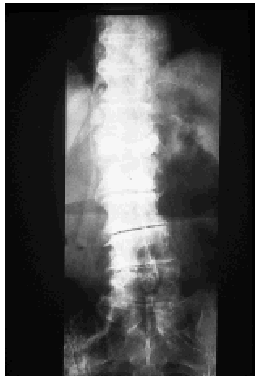
Fig. 4.--La radiografía de columna dorsolumbar demostró un aumento de densidad en algunos cuerpos vertebrales.
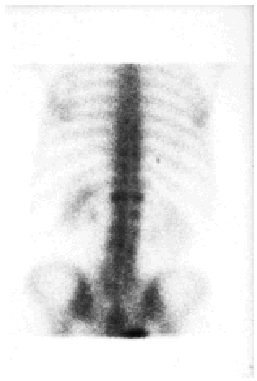
Fig. 5.--La gammagrafía ósea demostró un aumento de captación del trazador en la vértebra D12.
Se instauró tratamiento oral con hidroxicina a dosis de 75 mg diarios y prednisona a dosis inicial de 75 mg en pauta descendente, observándose una escasa respuesta en las lesiones cutáneas y una mejoría muy discreta de los dolores óseos y de la fiebre.
Hace unos 30 años la paciente había presentado lesiones cutáneas similares a las actuales, poco pruriginosas, también generalizadas, más intensas en miembros, respetando la cara, acompañadas de febrícula matutina. En aquel momento la paciente fue sometida a múltiples pruebas diagnósticas, incluidos un espectro electroforético, un estudio de la médula ósea y una biopsia de ganglio linfático, cuyos resultados fueron normales, y fue diagnosticada de urticaria crónica de acuerdo con una biopsia cutánea que reveló la presencia de un infiltrado perivascular superficial de linfocitos y neutrófilos. Las lesiones no respondieron al tratamiento antihistamínico y esteroideo por vía oral que se le administró en aquel momento, pero progresivamente fueron remitiendo de forma espontánea hasta hace unos dos años en que comenzó a presentar de nuevo la clínica antes mencionada.
DISCUSIÓN
La presencia de la erupción cutánea y del componente monoclonal IgM en el síndrome de Schnitzler son constantes, aunque se ha sugerido la posibilidad de ausencia de la primera a raíz de la publicación del caso de un paciente con dolores óseos, osteosclerosis y pico monoclonal IgM kappa sin manifestaciones cutáneas3. La presencia de una gammapatía monoclonal IgM se detecta en casi el 100% de los casos, siendo, por tanto, uno de los criterios que definen al síndrome. Sin embargo, en fases muy incipientes de la enfermedad es posible que el componente monoclonal sea indetectable por hallarse a concentraciones muy bajas2. Además se han publicado varios casos con gammapatías monoclonales combinadas IgM e IgA4 o IgM e IgG5 y se ha descrito el caso de un paciente con urticaria, fiebre, artralgia y elevación de la VSG en el contexto de una paraproteinemia monoclonal IgG aislada6.
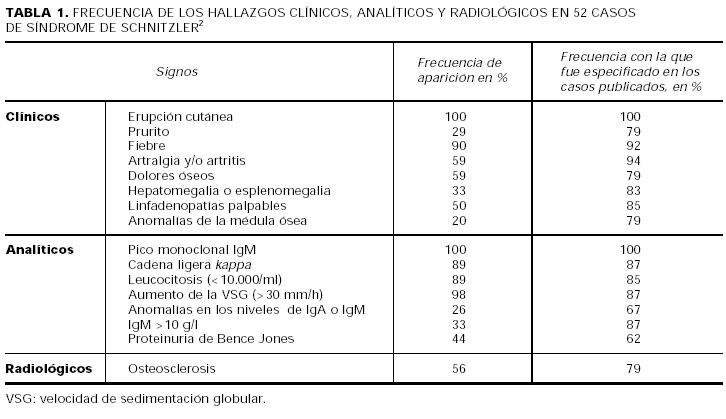
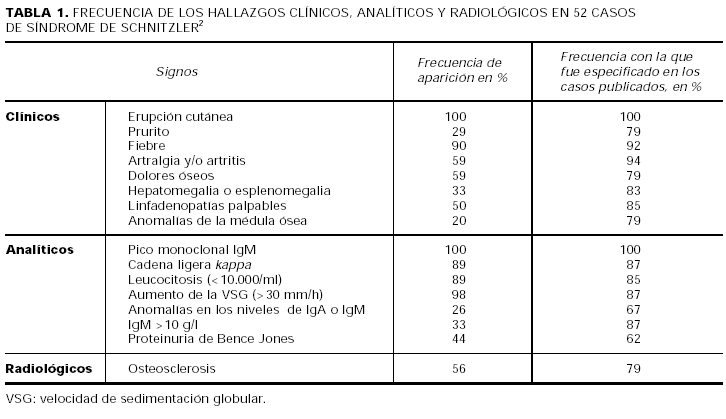
Recientemente Lipsker et al2 han publicado una revisión sobre el síndrome de Schnitzler. Estos autores han calculado la frecuencia relativa con la que aparece cada uno de los signos clínicos, analíticos y radiológicos en los casos de síndrome de Schnitzler publicados hasta la fecha en la literatura (tabla 1). De acuerdo con estas proporciones, han propuesto una serie de criterios para el diagnóstico de la enfermedad (tabla 2).

La erupción cutánea es habitualmente monomorfa y está constituida por numerosas maculopápulas o placas eritematorrosadas, ligeramente sobreelevadas, con tendencia a la confluencia y de un tamaño variable entre 0,5 y 2 ó 3 cm de diámetro. Es característico que las lesiones aparezcan diariamente para desaparecer sin dejar secuelas en un período que oscila entre 12 y 24 horas. El tronco y los miembros son las regiones afectadas con mayor frecuencia y la región cervicofacial generalmente está respetada. Habitualmente las lesiones son asintomáticas aunque se ha descrito la aparición de prurito en el 29% de los casos transcurridos 3 ó 4 años del inicio de las lesiones. El angioedema es un hallazgo excepcional. Otra característi-ca de la erupción cutánea asociada al síndrome de Schnitzler es su resistencia a los tratamientos2.
De Castro et al7 publicaron una revisión de los hallazgos histopatológicos presentes en el síndrome de Schnitzler estudiando 23 biopsias procedentes de 15 pacientes. En 13 biopsias de 9 pacientes se observó un infiltrado perivascular superficial constituido fundamentalmente por polimorfonucleares neutrófilos con leucocitoclastia leve o nula. Algunos casos presentaban también un infiltrado intersticial en dermis superficial y media de polimorfonucleares, siendo excepcional la presencia de eosinófilos. Los vasos de la dermis se hallaban intactos con leucocitos polimorfonucleares en su pared en algunas de las biopsias. La presencia de una vasculitis bien desarrollada sólo se detectó en dos biopsias procedentes de un mismo paciente que, además, tenía un déficit congénito de C4. En el 30% de los casos se han detectado depósitos de IgM alrededor de los vasos de la dermis superficial mediante técnicas de inmunofluorescencia y en algunos casos aislados depósitos de IgM a lo largo de la unión dermoepidérmica. Recientemente se ha demostrado que la interacción entre la IgM y la piel desempeña un importante papel en la fisiopatología de las lesiones cutáneas, mediante el desencadenamiento de una respuesta inflamatoria local2.
La fiebre, de tipo intermitente, con picos que pueden superar los 40° C, constituye otro de los signos cardinales del síndrome de Schnitzler y está presente en el 90% de los casos. Todavía no se han clarificado los mecanismos fisiopatológicos de la fiebre y del síndrome. Se ha descrito la presencia de anticuerpos anti-interleucina 1 (IL-1)8 en estos pacientes, pero esto no ha podido confirmarse en estudios posteriores. Todavía está por dilucidar si la expansión clonal de IgM es de naturaleza primaria o es secundaria a una estimulación antigénica continuada2.
Los signos musculoesqueléticos aparecen en el 89% de los casos en forma de dolores óseos en el 59% y/o de artralgia o artritis también en el 59% de los casos. Los dolores óseos suelen afectar al hueso ilíaco y a la tibia; le sigue en frecuencia la afectación del fémur, la columna vertebral, el antebrazo y la clavícula. Según la revisión más completa de los signos radiológicos del síndrome de Schnitzler9, el hallazgo más común es la osteocondensación en la radiografía simple, aunque se han publicado los casos de dos pacientes con lesiones osteolíticas10, 11. En el 40% de los casos se observan anomalías en la gammagrafía ósea que consisten en aumento de la captación del trazador. Cuando existe hiperostosis en la radiografía, la gammagrafía es anormal en la misma localización9. La causa de estas alteraciones es desconocida. Sólo se ha realizado biopsia ósea en 9 pacientes2, que fue normal en 3 de ellos y mostró una inflamación inespecífica en 5, en ocasiones asociada a osteoblastos hiperactivos, y un paciente tenía evidencia histológica de osteosclerosis.
En el 50% de los pacientes se palpan linfadenopatías, que suelen distribuirse en las regiones axilar e inguinal y ocasionalmente en la región laterocervical. Los ganglios pueden ser múltiples, persistentes y de tamaño variable entre 2 y 3 cm de diámetro. La biopsia de ganglio linfático demuestra la presencia de inflamación inespecífica. El 33% de los casos puede presentar hepatomegalia o esplenomegalia.
En el 80% de los pacientes el estudio de la médula ósea es normal en el momento del diagnóstico y en el 20% restante se han descrito infiltrados linfoplasmocitarios o plasmocitarios policlonales inespecíficos2.
El hallazgo de una VSG incrementada es un dato analítico constante en el síndrome de Schnitzler. Los niveles de complemento suelen estar normales o aumentados en estos pacientes. El 10% de los casos presenta trombocitosis y anemia reactiva. Se han publicado dos casos en los que la anemia era marcada y sintomática1, 12. En el 90% de los pacientes con síndrome de Schnitzler se ha descrito leucocitosis superior a los 10 x 103/mm3.
El diagnóstico diferencial debe plantearse con las entidades que figuran en la tabla 3. Tanto la enfermedad de Still del adulto como el síndrome de Schnitzler presentan erupción cutánea, fiebre, artralgias, linfadenopatías palpables, hepatoesplenomegalia y leucocitosis. Sin embargo, en la enfermedad de Still los niveles de ferritina están generalmente más elevados que en el síndrome de Schnitzler y el componente monoclonal IgM está ausente.

En la vasculitis urticariana hipocomplementémica la biopsia cutánea muestra la presencia de vasculitis, los niveles de complemento están disminuidos y con frecuencia se asocia a lupus eritematoso. Se ha descrito una deficiencia adquirida de inhibidor de esterasa C1, especialmente en el contexto de un linfoma y/o paraproteinemia. Esta entidad se distingue del síndrome de Schnitzler por la presencia de angioedema y de bajas concentraciones de C4 y niveles funcionales bajos de C1 inhibidor. En la crioglobulinemia los síntomas se desencadenan ante bajas temperaturas. Este dato y la presencia de crioglobulinas en sangre permite el diagnóstico diferencial entre esta entidad y el síndrome de Schnitzler. El síndrome hiper-IgD se caracteriza por la presencia de erupción cutánea que puede ser urticariana, fiebre periódica, artralgias, linfadenopatías cervicales, leucocitosis e incremento de la VSG; la presencia de altas concentraciones de IgD y la ausencia del componente monoclonal IgM lo distinguen del síndrome de Schnitzler. El eritema marginado reumático, que aparece en el 10% de los pacientes con fiebre reumática activa, es más frecuente en niños y en pacientes con afectación cardíaca, y se caracteriza por la presencia de lesiones cutáneas eritematosas y anulares que afectan fundamentalmente al tronco y que desaparecen espontáneamente en pocas horas y muestran tendencia a la recurrencia. El antecedente de una infección estreptocócica y el hallazgo de anticuerpos antiestreptocócicos lo distinguen del síndrome de Schnitzler. En el lupus eritematoso sistémico (LES) se ha descrito urticaria, fiebre y artralgias. Sin embargo, en el síndrome de Schnitzler no se observan los hallazgos clínicos y analíticos característicos del LES. El síndrome CINCA (síndrome crónico infantil neurológico, cutáneo y articular), también llamado NOMID (enfermedad neonatal multisistémica inflamatoria), comienza en la infancia y se caracteriza por afectación neurológica y deformidades articulares, ausentes en el síndrome de Schnitzler. El síndrome Muckle-Wells, caracterizado por brotes recidivantes de urticaria y fiebre desde la adolescencia, se diferencia del síndrome de Schnitzler por la historia familiar y la presencia de sordera y amiloidosis.
El curso del síndrome de Schnitzler es crónico, sin que se hayan podido demostrar remisiones espontáneas o inducidas por el tratamiento. El pronóstico está condicionado por la posibilidad de evolución a un proceso linfoproliferativo, bien linfoma, bien mieloma IgM o enfermedad de Waldenström, descrita en el 15% de los casos publicados2. El caso original descrito por Schnitzler et al en 1974 falleció de una infiltración linfoplasmocitaria difusa del hígado y la médula ósea 23 años después del comienzo de la enfermedad5. En la mayoría de los casos la progresión a un linfoma o enfermedad de Waldenström se produce a los 10-20 años después del inicio de los primeros signos de la enfermedad. Así, es posible que la proporción de casos que evolucionan a un trastorno linfoproliferativo esté infravalorada, ya que el tiempo de seguimiento de los casos publicados es, probablemente, insuficiente para predecir el pronóstico a largo plazo1. No existe ningún factor predic-tivo de la progresión a un trastorno linfoproliferati-vo. Por tanto, a todo paciente con el síndrome se le debe hacer un estudio basal que incluya inmunoelectroforesis de sangre y orina, cuantificación de inmunoglobulinas y estudio de médula ósea. Dos veces al año se repetirán las dos primeras determinaciones. Asimismo se realizará biopsia de ganglio linfático en todos aquellos casos en que se detecten linfadenopatías palpables.
El tratamiento de la erupción cutánea es desalentador. Los antiinflamatorios no esteroideos (AINE), en particular el ibuprofeno a dosis de 400 mg tres veces al día, son de primera elección, pero en la mayoría de los pacientes estos fármacos apenas mejoran el cuadro cutáneo. Se han utilizado antihistamínicos orales, colchicina, dapsona, hidroxicloroquina, cloroquina, esteroides, inmunosupresores, plasmaféresis, inmunoglobulinas intravenosas, quimioterapia y fototerapia con resultados variables. La respuesta al tratamiento de los síntomas sistémicos como la fiebre y los síntomas musculoesqueléticos es más favorable que la de las manifestaciones cutáneas. Se utilizan AINE asociados a veces a colchicina o dapsona en el caso de los dolores óseos. Los esteroides e inmunosupresores se reservan para aquellos casos en los que los síntomas sistémicos, esto es, la fiebre y los dolores osteoarticulares, sean incapacitantes y no respondan al tratamiento de primera línea con AINE. Otra indicación de los esteroides orales en los pacientes con síndrome de Schnitzler es el tratamiento de los raros casos de anemia intensa2.
En resumen, presentamos el caso de una paciente con urticaria crónica, oligosintomática y refractaria al tratamiento antihistamínico y esteroideo, con fiebre intermitente y dolores óseos, en el seno de una paraproteinemia monoclonal IgM kappa. Además, la paciente presentaba leucotrombocitosis, anemia inflamatoria, aumento de la VSG y osteosclerosis vertebral en la serie radiológica ósea. Se trata de una paciente que cumple los criterios clínicos, analíticos y radiológicos del síndrome de Schnitzler. Sorprende que en su historia previa la paciente revelara antecedentes de erupción cutánea similar aproximadamente 30 años atrás, que remitió espontáneamente, sin que pudiera demostrarse entonces gammapatía monoclonal en el proteinograma ni tampoco anomalías en el estudio de la médula ósea o en la biopsia de ganglio. Posteriormente y hasta hace dos años la paciente se ha encontrado libre de síntomas.


