

INTRODUCCION
El síndrome de Netherton es una rara enfermedad, trasmitida mediante herencia autosómica recesiva y definida por la tríada característica de dermatosis ictiosiforme, alteraciones del tallo piloso y trastornos inmunológicos 1,2. Recientemente se ha demostrado que la enfermedad se debe a mutaciones en el gen SPINK5 localizado en el cromosoma 5q32 3. Presentamos un caso de síndrome de Netherton, que es extremadamente infrecuente en España 2.
DESCRIPCION DEL CASO
Una niña de 12 años fue remitida a nuestra consulta por presentar eritema y descamación generalizados desde la infancia. Como antecedentes personales, la paciente había presentado un fenotipo de bebé colodión al nacimiento y posteriormente diagnosticada de eritrodermia ictiosiforme congénita no ampollosa mediante una biopsia practicada al mes de vida. Desde entonces, la paciente presentaba un enrojecimiento difuso de la piel junto con descamación. Además, desde los pocos meses de vida, desarrolló lesiones de eritema, edema, vesiculación y costras, de curso en brotes e intensamente pruriginosas, que habían sido diagnosticadas de dermatitis atópica y tratadas con éxito con corticoides tópicos. Entre las analíticas realizadas previas a nuestra valoración destacaba una cifra muy elevada de IgE (21.400 U/ml).
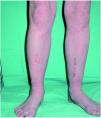
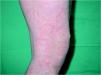
A la exploración física, la niña mostraba un desarrollo normal, y en la piel presentaba una eritrodermia moderada. Sobre dicha eritrodermia se apreciaban áreas de descamación de formas policíclicas y serpiginosas que, según la paciente, cambiaban de forma y de tamaño progresivamente. En la periferia de las mismas se apreciaba un doble borde descamativo (figs. 1 y 2). El cabello era escaso, frágil y quebradizo. Un tricograma mostró pelos con bulbos anágenos normales en su mayoría y no se apreciaron anomalías en el tallo del pelo. En muestras de pelos de la ceja se observó en la mayoría de los tallos pilosos la presencia de fracturas transversales, donde la parte distal se invaginaba en la parte proximal (fig. 3). El resto de la exploración física fue normal. Las analíticas realizadas, incluyendo hemograma y bioquímica sérica no ofrecieron resultados anómalos. Se instauró tratamiento con acitretina oral, 10 mg/día, con buena respuesta clínica de la piel.
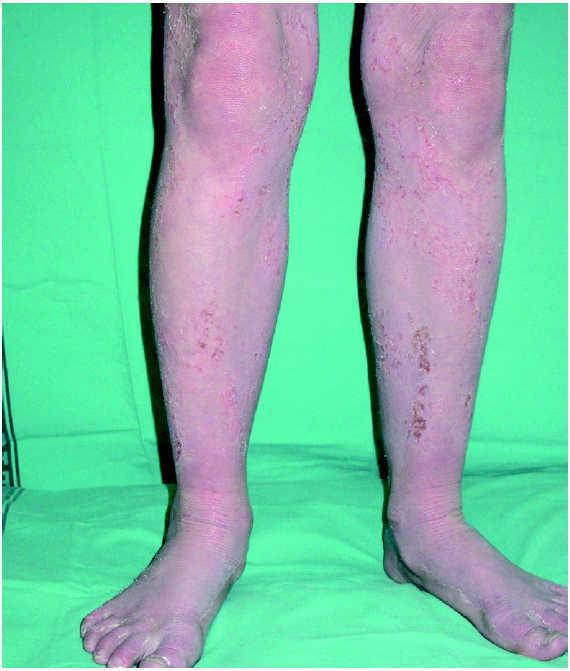
Fig. 1.--Eritrodermia con descamación.
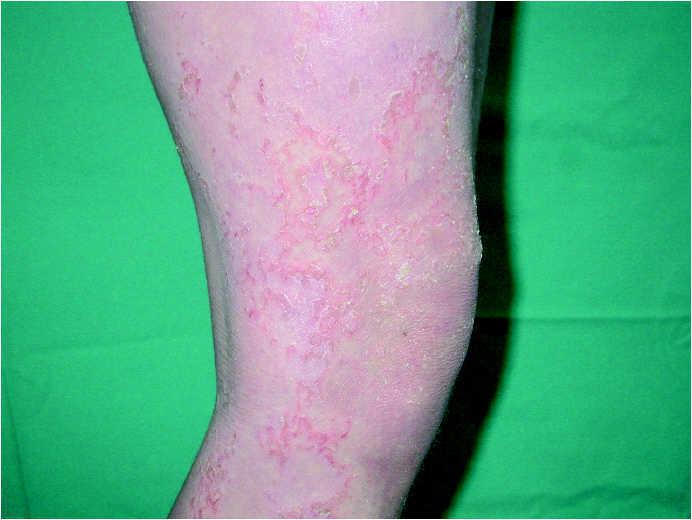
Fig. 2.--Lesiones de ictiosis lineal circunfleja.
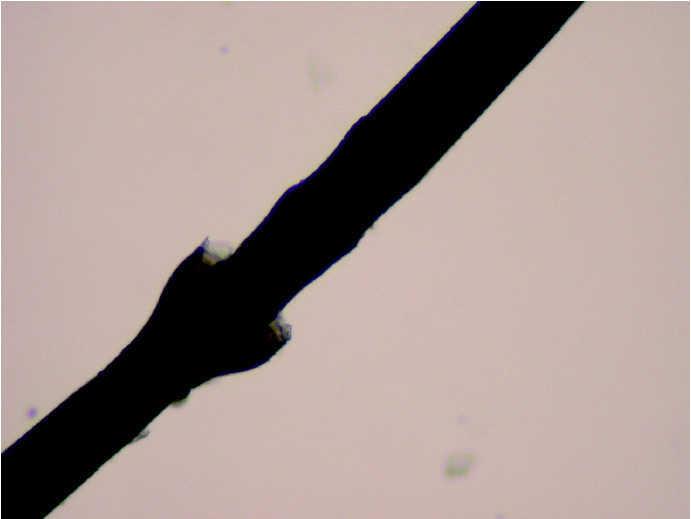
Fig. 3.--Tricorrexis invaginata en pelos de cejas.
DISCUSION
El síndrome de Netherton es una enfermedad rara, de herencia autosómica recesiva 4,5. Recientemente se ha demostrado como responsable de esta entidad se debe a mutaciones en el gen SPINK5 localizado en el cromosoma 5q32 3-5. Dicho gen codifica una proteína, inhibidora de la serinproteasa, llamada LEKTI, de tal forma que al no producirse dicha proteína, o al hacerlo de forma defectuosa, se produce una situación proteolítica incontrolada que provoca alteraciones en la ultraestructura del estrato córneo de la epidermis y rotura de los puentes disulfuro en el tallo piloso, produciendo la clínica cutánea y capilar característica 6. Los trastornos inmunológicos también se podrían explicar por la mutación de dicho gen, que produciría alteraciones en la diferenciación de los linfocitos T y producción aumentada de IgE 7.
La enfermedad suele empezar al nacimiento o poco después del mismo, de forma inespecífica, con eritrodermia y descamación, lo que produce frecuentes errores diagnósticos. El 18 % de una serie de 51 pacientes con eritrodermia neonatal o infantil fue finalmente diagnosticado de síndrome de Netherton 8. Como estos pacientes tienen una importante alteración de la función barrera de la piel, pueden presentar complicaciones en el período neonatal en forma de deshidratación, hipernatremia, fallo en el desarrollo y alteraciones en la termorregulación, entre otros 1,9,10. Los pacientes pueden presentar una eritrodermia ictiosiforme congénita no ampollosa, aunque la mayoría, acaban desarrollando unas lesiones más específicas y definidas como ictiosis lineal circunfleja 2. Estas lesiones consisten en placas eritematosas de formas policíclicas o serpiginosas que cambian de forma y tamaño progresivamente y que presentan un doble borde descamativo.
En cuanto a la clínica del tallo piloso, las alteraciones suelen desarrollarse en la infancia y la mayoría de pacientes se quejan de pelo escaso, frágil y de crecimiento lento. Aunque se pueden observar múltiples trastornos en la microscopia óptica, tales como pili torti, tricorrexis nodosa o moniletrix, se considera patognomónica del síndrome de Netherton una alteración del tallo piloso consistente en fracturas transversales del mismo donde la parte más distal se invagina en la proximal, denominada tricorrexis invaginata 9,10. La tricorrexis invaginata puede encontrarse tanto en el pelo del cuero cabelludo como en cejas, pestañas o vello axilar o púbico 8. La mayoría de los autores insiste en la necesidad de tomar muestras repetidas hasta encontrar los hallazgos descritos 8, ya que éstos pueden estar ausentes en los cabellos y presentes en cejas o pestañas. Las alteraciones del tallo piloso suelen mejorar con la edad.
Completando la tríada característica del síndrome de Netherton se encuentra la diátesis atópica, aunque muchos autores prefieren referirse a alteraciones inmunológicas en general, con reacciones alérgicas, elevación importante de IgE, infecciones frecuentes, urticaria o shock 1,2,4,5.
Para el diagnóstico de síndrome de Netherton puede ser suficiente con las manifestaciones clínicas, pero en ocasiones, sobre todo durante el primer año de vida, éstas no son muy evidentes y el diagnóstico se retrasa o se establece de forma errónea 8. La histopatología muestra un patrón psoriasiforme inespecífico con hiperqueratosis paraqueratósica, acantosis y papilomatosis 6,9. Los hallazgos ultraestructurales son más específicos y consisten en una exocitosis prematura de los cuerpos lamelares y alteraciones en la ultraestructura del estrato córneo 11. Recientemente se ha demostrado mediante inmunohistoquímica la ausencia de la proteína LEKTI en el estrato granuloso de la epidermis en los pacientes con síndrome de Netherton 10.
El tratamiento dependerá, en primer lugar, de la presencia de complicaciones en el período neonatal; en tal caso puede ser necesario el ingreso de estos pacientes en unidad de cuidados neonatales. Para las lesiones cutáneas puede ser necesario el empleo de emolientes, corticoides tópicos, queratolíticos e incluso de retinoides orales 1. Es importante destacar que, por la alteración de la función barrera de estos pacientes, se han descrito casos de absorción sistémica significativa con el uso tópico de tacrolimus y corticoides para su dermatitis atópica 12.
En cuanto al pronóstico de estos pacientes, se han descrito casos de desarrollo de múltiples neoplasias cutáneas y se ha señalado como etiología a las alteraciones inmunológicas provocadas por la mutación del gen SPINK57.
Declaración de conflicto de intereses Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Antonio Torrelo.
Servicio de Dermatología.
Hospital Niño Jesús.
Avda. Menéndez Pelayo, 65.
28009 Madrid. España.
atorrelo@aedv.es
Recibido el 27 de marzo de 2006.
Aceptado el 24 de abril de 2006.


