







El síndrome de Gorlin es una enfermedad infrecuente de herencia autosómica dominante producida por mutaciones en genes de la vía de señalización Sonic Hedgehog, entre los que destaca PTCH1. Se caracteriza por el desarrollo de múltiples carcinomas basocelulares en edades tempranas, que pueden ir asociados a otras manifestaciones cutáneas como pits palmoplantares, o a manifestaciones extracutáneas, entre las que destacan los queratoquistes odontogénicos y el meduloblastoma. El papel del dermatólogo es importante en la sospecha de este síndrome, pero suele ser necesario un equipo multidisciplinar en el diagnóstico, seguimiento y en el tratamiento de estos pacientes. El tratamiento dermatológico puede ser complicado debido al alto número de carcinomas basocelulares y a su extensión. En los últimos años se han desarrollado nuevos fármacos que inhiben la vía Sonic Hedgehog y parecen prometedores para estos pacientes, aunque su eficacia está limitada por los efectos secundarios y la creación de resistencias.
Gorlin syndrome is a rare autosomal dominant disease caused by mutations in the sonic hedgehog signaling pathway. Of particular importance is the PTCH1 gene. The disease is characterized by the development of multiple basal cell carcinomas at young ages. These tumors may present with other skin manifestations such as palmoplantar pits and with extracutaneous manifestations such as odontogenic keratocysts and medulloblastoma. Although the dermatologist may be key for recognizing clinical suspicion of the syndrome, a multidisciplinary team is usually necessary for diagnosis, treatment, and follow-up. Skin treatment may be complicated due to the large number of basal cell carcinomas and the extent of involvement. In recent years, new drugs that inhibit targets in the sonic hedgehog pathway have been developed. Although these agents appear promising options for patients with Gorlin syndrome, their efficacy is limited by adverse effects and the development of resistance.
El síndrome de Gorlin (SG) o síndrome del carcinoma basocelular nevoide (OMIM: 109400) es una enfermedad genética de herencia autosómica dominante que predispone a la presencia de defectos del desarrollo y al desarrollo de neoplasias, entre los que destacan carcinomas basocelulares (CBC) múltiples1. La patogenia molecular de este síndrome se ha relacionado con el gen patched 1 (PTCH1), que codifica para el receptor transmembrana PTCH1, implicado en la vía de señalización Sonic Hedgehog (SHH)2–4. Las posibilidades terapéuticas de estos enfermos han avanzado recientemente con la introducción del inhibidor de SHH vismodegib, indicado en el tratamiento de CBC metastásico, recurrente o localmente avanzado5.
EpidemiologíaEl SG tiene una prevalencia variable según las series publicadas, de entre 1/308276 y 1/2560007. Farndon et al. establecieron una prevalencia mínima de esta enfermedad de 1/57000 habitantes, y estimaron que uno de cada 200 pacientes con uno o más CBC tienen SG8.
La esperanza de vida de los enfermos de SG es 73,4 años, significativamente más reducida que la de la población general, que es de aproximadamente 80 años9. La causa más importante de muerte prematura en estos pacientes es por meduloblastoma10.
Patogenia molecularEl SG es una enfermedad genética de herencia autosómica dominante, con alta penetrancia y expresividad variable11. Se produce por pérdida de heterocigosidad del gen supresor tumoral PTCH1, mapeado en el cromosoma 9q22.32. PTCH1 forma parte de la vía de señalización SHH, por lo que mutaciones en este gen conllevarían una sobreexpresión de la ruta SHH4.
La vía de señalización SHH fue descrita por primera vez en Drosophila12 y es esencial durante el desarrollo, ya que interviene en la polaridad tisular y la población de células madre13. En los mamíferos está compuesta por cuatro elementos principales (fig. 1):
- 1.
Ligandos Hegdehog (LHH) de PTCH 1: Sonic Hedgehog, Indian Hedgehoh y Desert Hedgehog.
- 2.
El receptor PTCH1.
- 3.
La proteína transductora de señales smoothened (SMO).
- 4.
Los factores de transcripción Gli1, Gli2, Gli313.

PTCH1 inhibe constitutivamente la actividad de la proteína SMO. La unión de los LHH a PTCH1 suprime la actividad inhibitoria de PTCH1 sobre SMO. Una vez liberada, SMO se trasloca al extremo del cilio primario para ejercer su función, que tiene como resultado la activación de los factores de transcripción Gli14,15. Las proteínas Gli promueven la transcripción de genes implicados en el aumento de la supervivencia celular y mitosis16. En vertebrados existen 3 proteínas Gli. GLI 1 y GLI2 tienen función activadora, mientras que GLI3 impide la trascripción de los genes diana14, entre los que se encuentran los propios genes Gli y PTCH1. Se ha demostrado además una relación entre la vía SHH y otras rutas de señalización como la del factor de crecimiento epidérmico, factor de crecimiento insulínico, factor de crecimiento transformante beta (TGF-β), mammalian Target of Rapamycin (mTOR)/S6K1, receptor proteína quinasa c 1, notch, wnt/β-catenina y fosfoinosítido-3-quinasa (PI3K/Akt), de modo que todas ellas modulan la patogenia del cáncer4,13,14,17.
PTCH1 puede hallarse mutado entre el 50-85% de los enfermos con SG18,19 y en un 20-30% estas mutaciones son de novo20. Con menor frecuencia se encuentran mutados otros genes de la vía SHH21, entre los que destacan Suppressor of fused (SUFU)22, PTCH2, SMO o GLI23. El gen alterado con mayor frecuencia tras PTCH1 es SUFU, y debe ser investigado en aquellos pacientes con test genético negativo para PTCH111. Se ha relacionado la presencia de una mutación inactivadora de SUFU con una menor penetrancia y con un menor número de criterios diagnósticos mayores. Además, estos pacientes tienen mayor riesgo de meduloblastoma y no presentan queratoquistes odontogénicos22. Las mutaciones en PTCH2 son raras en pacientes con SG, pero estos enfermos tienen un fenotipo más leve23. Así mismo, los casos de mosaicismo somático producido por una mutación en una etapa precoz del desarrollo embrionario son infrecuentes20.
Los pacientes con SG nacen en su mayoría con una mutación heredada en uno de los alelos de PTCH1, que representa un alelo nulo y codifica una proteína truncada. Para que se produzca la enfermedad se debe cumplir el fenómeno del «doble hit» de Knudson. El primer «hit» o evento sería la mutación heredada y el segundo evento se correspondería con una mutación adquirida sobre el alelo sano del gen, que puede producirse por factores externos como la luz UV21.
Manifestaciones cutáneas y extracutáneas del síndrome de Gorlin. Criterios diagnósticosHasta la fecha no se ha conseguido establecer una correlación entre el fenotipo y el genotipo de estos pacientes11. Se han descrito distintas clasificaciones de criterios diagnósticos de la enfermedad, que incluyen manifestaciones cutáneas y extracutáneas20,24–28. En 2011 se publicó un documento de consenso con nuevos criterios diagnósticos en el que se incluyó por primera vez el estudio molecular (tabla 1). Para poder llegar al diagnóstico de SG serían necesarios dos criterios mayores, un criterio mayor y dos menores, o un criterio mayor y la confirmación molecular26. Sin embargo, no se ha evaluado todavía la sensibilidad y especificidad de las diferentes propuestas de criterios diagnósticos11.
Criterios diagnósticos de síndrome de Gorlin
| Criterios diagnósticos |
|---|
| Criterios mayores |
| CBC en <20 años o número excesivo de CBC para la exposición solar y fototipo |
| Queratoquistes mandibulares en <20 años |
| Pits palmoplantares |
| Calcificación lamelar de la hoz cerebral |
| Meduloblastoma |
| Familiar de primer grado con SG |
| Otras anomalías esqueléticas y cambios radiológicos (anomalías vertebrales, cifoescoliosis, 4 metacarpiano corto, polidactilia postaxial) |
| Criterios menores |
| Anomalías costales |
| Macrocefalia |
| Fibromas ováricos o cardiacos |
| Anomalías oculares (estrabismo, hipertelorismo, catarata congénita, glaucoma, coloboma) |
| Labio o paladar hendido |
| Quistes linfomesentéricos |
Para establecer el diagnóstico son necesarios dos criterios mayores, un criterio mayor y dos menores o un criterio mayor y la confirmación molecular. Fuente: Bree et al.26.
- -
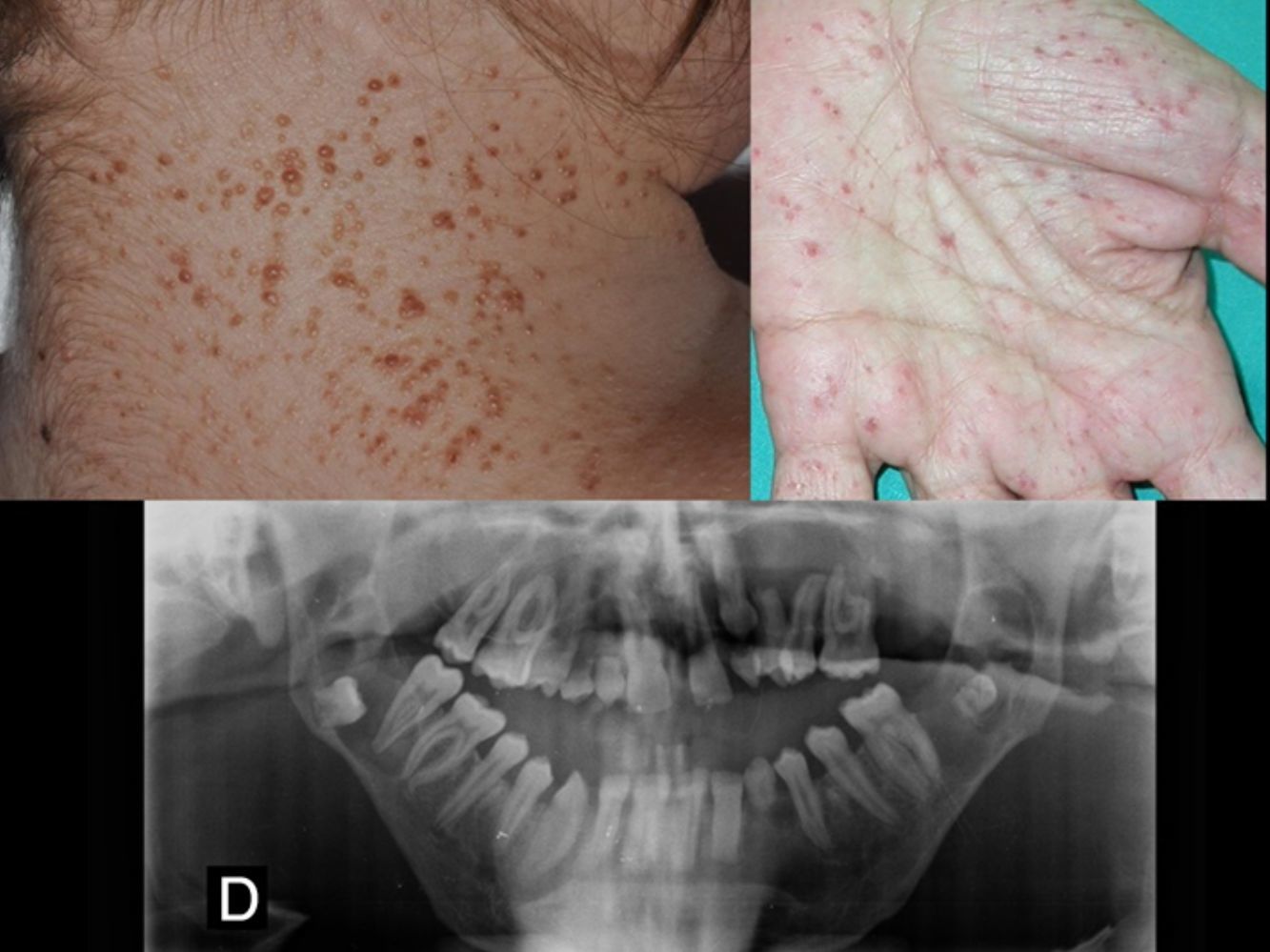
Carcinomas basocelulares (fig. 2). Pueden aparecer en edades tempranas de la vida y ser múltiples26,28. Los CBC en el SG pueden afectar tanto a áreas fotoexpuestas como no fotoexpuestas, con un ligero predominio de las expuestas1. Las localizaciones más frecuentes en hombres son el tercio superior de espalda, extremidades superiores y la zona M facial, mientras que en las mujeres son el cuero cabelludo, espalda y extremidades inferiores. La proporción de cada subtipo histológico de CBC es similar al de los pacientes con CBC esporádico29.
- -
Pits palmoplantares (fig. 3). Son una manifestación frecuente y se encuentran entre el 70-87% de los enfermos24,27. Alrededor del 30-65% de los pacientes con SG presentan pits palmoplantares antes de 10 años de edad y la mayoría los habrán desarrollado a los 15 años10. Son depresiones puntiformes, de 2-3mm de diámetro, que aparecen en palmas y en plantas y más raramente en dorso o laterales de dedos y pliegues interdigitales27. Histológicamente son áreas de hipoqueratosis con hipogranulosis variable, paraqueratosis e hiperplasia de células basales, que forman empalizada en la periferia30.
- -
Otras manifestaciones cutáneas. Los nevus melanocíticos múltiples son frecuentes. Se ha descrito también una mayor incidencia de quistes de millium en párpado inferior y frente y quistes epidermoides en tronco10.


- -
Queratoquistes mandibulares u odontogénicos (fig. 4). Se encuentran entre el 74-90% de los pacientes con SG11,27. Pueden iniciarse a los 4-5 años y aparecen antes de los 20 años en el 75% de los casos27, pero su desarrollo es raro en mayores de 30 años. Son asintomáticos, se localizan en ambos lados de la mandíbula y habitualmente son múltiples10. Ocasionalmente pueden malignizar a ameloblastomas11 y a carcinoma epidermoide21.
 Ortopantomografía, donde se aprecian queratoquistes engoblando varios molares. b) Corte axial de tomografía computerizada en el que se observan queratoquistes odontogénicos maxilares. c) Corte coronal de tomografía computerizada, donde se visualizan queratoquistes maxilares que comprimen los senos paranasales. Cortesía de la Dra. E. García Esparza.") Figura 4.
Figura 4.Queratoquistes odontogénicos en paciente con síndrome de Gorlin. a) Ortopantomografía, donde se aprecian queratoquistes engoblando varios molares. b) Corte axial de tomografía computerizada en el que se observan queratoquistes odontogénicos maxilares. c) Corte coronal de tomografía computerizada, donde se visualizan queratoquistes maxilares que comprimen los senos paranasales. Cortesía de la Dra. E. García Esparza.
- -
Manifestaciones no tumorales del sistema nervioso central. La más frecuente es la calcificación de la hoz del cerebro, presente en el 65-79% de los enfermos24,27,28. Este hallazgo no se detecta en la infancia precoz31 pero sí a partir de la adolescencia28. Otras alteraciones más raras son la calcificación de la tienda del cerebelo, del ligamento petroclinoide y del diafragma sellae32.
- -
Anomalías faciales. Se ha detectado macrocefalia relativa e hipertelorismo27,33,34, asociado en muchos casos a telecantus27. Se ha descrito mayor frecuencia de abombamiento de los huesos frontal, biparietal o temporal21.
- -
Alteraciones en vértebras, costillas y escápula. Entre el 38-49% de los pacientes presentan anomalías costales27,35, siendo la más común la presencia de costillas bífidas28. Otras alteraciones costales son las costillas separadas, fusionadas, ausentes, costillas cervicales y costillas rudimentarias. Las malformaciones congénitas vertebrales son frecuentes, sobre todo la escoliosis28,36 y el proceso espinoso bífido. Son hallazgos más raros la espina bífida oculta, hemivértebras, cuerpos vertebrales elongados, espondilolistesis y fusión de cuerpos vertebrales28. La elevación congénita de la escápula o enfermedad de Sprengel puede ser detectada entre el 11-22% de los pacientes28,35.
- -
Alteraciones en miembros. Las anomalías más relevantes son las radiolucencias con forma de llama en radiografías de manos, presentes en el 30% de los enfermos. Se ha descrito una mayor tasa de defectos de modelaje en manos y pies, sindactilia, polidactilia y de acortamiento del quinto metacarpiano28.
- -
Alteraciones oculares y auditivas. Además del hipertelorismo, se ha notificado una mayor prevalencia de exoftalmus, nistagmo rotatorio, estrabismo interno, cataratas congénitas, coloboma del iris y coroides y microftalmia10,24,37. Entre las anomalías otológicas destacan la otosclerosis, la sordera de conducción y el angulamiento posterior de las orejas33.
 Ortopantomografía, donde se aprecian queratoquistes engoblando varios molares. b) Corte axial de tomografía computerizada en el que se observan queratoquistes odontogénicos maxilares. c) Corte coronal de tomografía computerizada, donde se visualizan queratoquistes maxilares que comprimen los senos paranasales. Cortesía de la Dra. E. García Esparza.")
- -
Meduloblastoma. Es el tumor maligno cerebral más frecuente en la población pediátrica38. Se desarrolla en un 1-5% de los enfermos con SG24,27 por alteración de la vía SHH, y muchas veces es la primera manifestación de esta enfermedad38. Se detecta de media a los 2 años de edad, mientras que en la población general aparece entre los 7-8 años de edad10,27,38. El pronóstico de estos meduloblastomas es intermedio, con tasas de supervivencia global entre 60-80%39. El tratamiento estándar del meduloblastoma es la cirugía en combinación con radioterapia y quimioterapia. En pacientes con SG el tratamiento radioterápico puede conducir a la aparición de CBC y otros tumores cerebrales sobre el área de radiación, por lo que es necesario identificar a estos pacientes para optimizar el tratamiento adyuvante38.
- -
Fibromas cardiacos y ováricos. Los fibromas cardiacos son infrecuentes, están presentes en el nacimiento o al poco de nacer11, y pueden causar arritmias graves e insuficiencia cardiaca23. Los fibromas ováricos son generalmente un hallazgo incidental. Son asintomáticos y su transformación maligna es infrecuente, pero pueden causar problemas de fertilidad11.
- -
Otros tumores. Se han descrito meningiomas asociados a SG hasta en el 5% de los pacientes28, en algunos casos sobre un área tratada con radioterapia27. Son más raros tumores craneales como astrocitoma, craneofaringioma y oligodendroglioma, así como tumores sólidos y hematológicos10, entre los que destaca el rabdomioma fetal23,40,41.
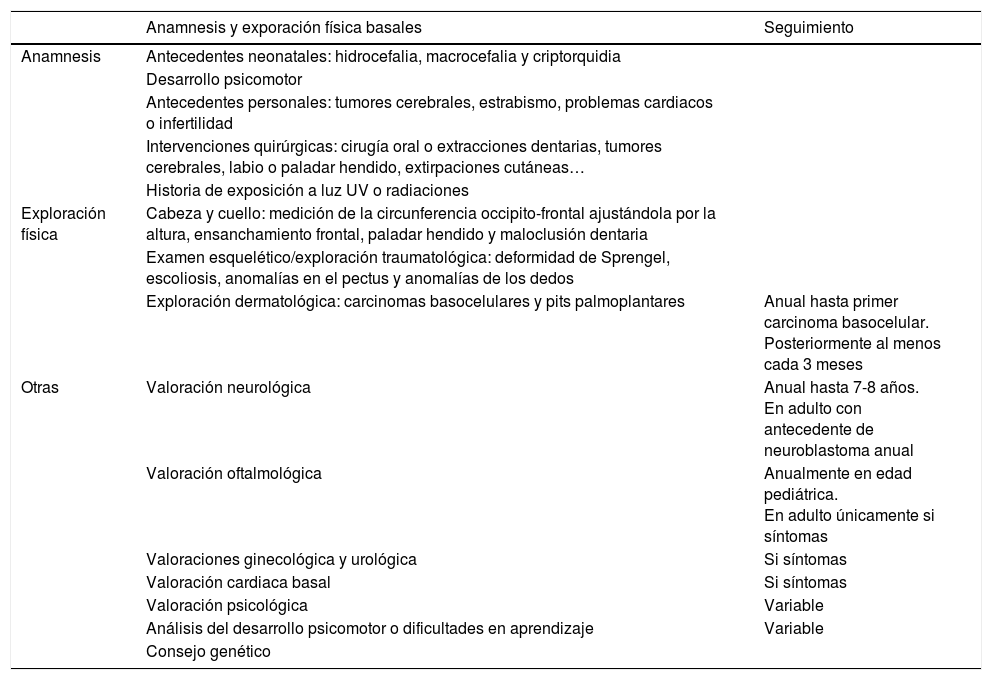
Existen escasas publicaciones en la literatura médica sobre qué pruebas diagnósticas deben ser realizadas en los pacientes con SG. El documento de consenso de 2011 enumera los datos clave de la historia clínica, de la exploración física y las exploraciones complementarias recomendadas para el diagnóstico y seguimiento de estos enfermos (tablas 2 y 3). Es importante minimizar las radiaciones ionizantes y si es posible se aconseja emplear la resonancia magnética o la ecografía26.
Anamnesis y exporación física en pacientes con síndrome de Gorlin
| Anamnesis y exporación física basales | Seguimiento | |
|---|---|---|
| Anamnesis | Antecedentes neonatales: hidrocefalia, macrocefalia y criptorquidia | |
| Desarrollo psicomotor | ||
| Antecedentes personales: tumores cerebrales, estrabismo, problemas cardiacos o infertilidad | ||
| Intervenciones quirúrgicas: cirugía oral o extracciones dentarias, tumores cerebrales, labio o paladar hendido, extirpaciones cutáneas… | ||
| Historia de exposición a luz UV o radiaciones | ||
| Exploración física | Cabeza y cuello: medición de la circunferencia occipito-frontal ajustándola por la altura, ensanchamiento frontal, paladar hendido y maloclusión dentaria | |
| Examen esquelético/exploración traumatológica: deformidad de Sprengel, escoliosis, anomalías en el pectus y anomalías de los dedos | ||
| Exploración dermatológica: carcinomas basocelulares y pits palmoplantares | Anual hasta primer carcinoma basocelular. Posteriormente al menos cada 3 meses | |
| Otras | Valoración neurológica | Anual hasta 7-8 años. En adulto con antecedente de neuroblastoma anual |
| Valoración oftalmológica | Anualmente en edad pediátrica. En adulto únicamente si síntomas | |
| Valoraciones ginecológica y urológica | Si síntomas | |
| Valoración cardiaca basal | Si síntomas | |
| Valoración psicológica | Variable | |
| Análisis del desarrollo psicomotor o dificultades en aprendizaje | Variable | |
| Consejo genético |
Pruebas complementarias en pacientes con síndrome de Gorlin
| Pruebas complementarias basales | Seguimiento | |
|---|---|---|
| Despistaje de criterios diagnósticos | Radiografías simples: parrilla costal, columna vertebral, manos y pies, pelvis (♀) | |
| Ortopantomografía | Al menos anual desde 8 años o que el niño colabore hasta primer queratoquiste. Desde entonces cada 6 meses hasta 21 años o dos años sin queratoquistes. En adultos si síntomas | |
| Ecografía ginecológica | Repetir si síntomas | |
| Ecografía cardiológica | Repetir si síntomas | |
| Estudio genético | ||
| Paciente pediátrico | Resonancia nuclear magnética craneal | Anual hasta 7-8 años |
| Paciente adulto | Radiología simple craneal (a partir de adolescencia) |
El estudio genético constituye uno de los criterios mayores propuestos en el consenso de 201126. Solo se consigue demostrar mutación en PTCH en alrededor del 60-80% de los pacientes20,21, por lo que se recomienda reservarlo para tres supuestos: en el diagnóstico prenatal si se conoce la mutación en la familia; para un diagnóstico de confirmación en pacientes que presentan algunos signos de enfermedad pero no cumplen los criterios diagnósticos; o como test predictivo en individuos con riesgo de presentar la enfermedad, que no cumplen criterios diagnósticos pero tienen un familiar afectado26.
Pruebas complementarias (tabla 3)Es importante tener en cuenta la edad de aparición de las manifestaciones de la enfermedad para elegir las pruebas diagnósticas más adecuadas.
Se recomiendan radiografías simples de parrilla costal y columna vertebral para descartar varios de los criterios menores de la enfermedad presentes desde el nacimiento27. Las radiografías de manos y pies también pueden detectar cambios en la infancia28. La radiografía craneal para el diagnóstico de calcificación de la hoz cerebral puede ser de utilidad en el paciente adulto11,28, teniendo en cuenta que este hallazgo se produce a partir de la adolescencia.
Los queratoquistes odontogénicos aparecen a partir de los 4 años de edad, por lo que se recomienda ortopantomografía digital anual en los pacientes con sospecha o diagnóstico confirmado, desde que el niño colabore hasta la aparición del primer queratoquiste26,27. Otros autores recomiendan el inicio del cribado a los 8 años11, y debe repetirse cada 6 meses hasta que el paciente cumpla 21 años. En adultos las ortopantomografías pueden repetirse anualmente si el paciente presenta síntomas26.
El riesgo de meduloblastoma es mayor entre los 2 y 3 años de edad, pero puede aparecer hasta los 7 años. Se aconseja seguimiento neurológico y resonancia magnética cerebral anual hasta los 7-8 años de edad. En el paciente adulto con antecedente de meduloblastoma se recomienda una resonancia cerebral basal y seguimiento anual en Neurología26.
Se aconseja una ecografía ovárica en mujeres asintomáticas en la menarquía o a los 18 años, o antes si refieren síntomas26,27. Si el diagnóstico de SG es en una mujer adulta, deberá realizarse ecografía ginecológica basal26. Puede ser necesario el consejo prenatal y seguimiento del embarazo para detección precoz de signos de la enfermedad en el feto24,26. Los fibromas cardiacos son infrecuentes y asintomáticos, pero puede plantearse un ecocardiograma en el primer año de vida para detectarlos11,26.
Además de todas estas consideraciones, puede ser necesario consejo genético y ayuda psicológica26.
PrevenciónLas medidas de prevención en pacientes con SG incluyen modificaciones en el estilo de vida y tratamiento médico. Se recomienda no exponer a los pacientes a radiaciones ionizantes, la protección solar debe ser estricta y se aconsejan suplementos de vitamina D42. Entre los fármacos sistémicos para la quimioprevención de CBC se han propuesto los retinoides. El más empleado es la isotretinoína oral, que debería pautarse a altas dosis durante largos periodos para conseguir resultados, lo que deriva en una intensificación de los efectos secundarios. Además, las lesiones recurren al suspender el fármaco33,43. Se ha ensayado también la quimioprevención con tazaroteno tópico sin haberse encontrado un efecto preventivo o terapéutico44. Algunos autores han sugerido también el uso de terapia fotodinámica (TFD)45,46 pero su utilidad no ha sido bien estudiada.
Tratamiento del carcinoma basocelular en pacientes con síndrome de GorlinLos pacientes con SG deben recibir un tratamiento multidisciplinar debido a las diferentes manifestaciones de la enfermedad11,26. En este trabajo nos centraremos en las modalidades terapéuticas para los CBC en estos enfermos, que se basan en el control local de la enfermedad47 con la mayor preservación de la función y proporcionando el mejor resultado cosmético posible48. Hemos dividido las modalidades de tratamiento en tres grupos: tratamiento quirúrgico, no quirúrgico y basado en la patogenia molecular.
Tratamiento quirúrgicoLa extirpación quirúrgica convencional es un método comúnmente empleado para los pacientes con SG pero que ha sido poco estudiado en la literatura médica. No hay estudios hasta la fecha sobre los resultados de esta técnica ni sobre las tasas de recidivas en estos pacientes. Los márgenes de seguridad recomendados son de al menos 4mm48.
La cirugía micrográfica de Mohs está indicada como tratamiento en el SG21,49, sobre todo en tumores de alto riesgo, recurrentes, que afecten al área facial u otras áreas de riesgo50. Mediante esta técnica la tasa de curación a 5 años de CBC primarios es del 99%, y en CBC recurrentes del 94,4%51, pero no existen datos en pacientes con SG.
Se ha recomendado tanto el empleo de curetaje y electrocoagulación como de la crioterapia en algunos casos de CBC primario de pequeño tamaño, bien definidos e histológicamente no agresivos, que asienten en áreas de bajo riesgo de recurrencia como el tronco o extremidades36,50,51 En estos casos, la tasa de curación a los 5 años es entre el 92-97% para los CBC esporádicos48,51, pero no existen estudios en SG. Ambas opciones podrían ser valoradas en casos de múltiples CBC y lesiones extensas42.
Se han publicado casos de tratamiento de CBC con láser de CO2 ablativo, que permitiría tratar varios CBC superficiales que asienten sobre un mismo área52–54.
Tratamiento no quirúrgicoLa TFD es un tratamiento conocido y ampliamente empleado en el tratamiento de CBC. En Europa se utilizan los fotosensibilizantes ácido aminolevulínico y metilaminolevulinato55. Se ha observado una tasa de desaparición completa de CBC esporádicos superficiales primarios tratados con TFD con metilaminolevulinato a los 3 meses del 92-97%, con tasas de recurrencia del 9% al año y del 22% a los 5 años56,57. La TFD con metilaminolevulinato se considera efectiva y segura en pacientes con SG, permitiendo el tratamiento simultáneo de múltiples CBC y de grandes áreas corporales. Este tratamiento se recomienda para los CBC superficiales de cualquier tamaño y CBC nodulares menores de 2mm de espesor58.
La quimioterapia tópica con 5-Fluorouracilo (5FU) al 5% está aprobada para el tratamiento de CBC en tumores de bajo riesgo extrafaciales y de espesor fino59. Se han publicado pacientes con SG con resultados contradictorios60,61, aunque puede resultar útil en combinación con crioterapia62 o tretinoína tópica63. Así mismo, se ha comunicado un enfermo con SG tratado satisfactoriamente con capecitabina oral, un profármaco del fluorouracilo, consiguiendo regresión de múltiples CBC y con buena tolerancia64.
La inmunoterapia con imiquimod 5% en crema está aprobada para la utilización en CBC superficiales, empleándola una vez al día, 5 días por semana durante 6 semanas59,65. No está indicado en CBC morfeiforme, infiltrativo, recurrente, o en lesiones en la cabeza47. En CBC esporádicos se ha objetivado una tasa de curación histológica del 82-90% a las 12 semanas65,66 y una tasa de recurrencia del 20,6% a los dos años66. Se han descrito pacientes con SG tratados con imiquimod 5% durante periodos variables de 6 a 14 semanas con buenos resultados67–70, pero no se dispone de datos a largo plazo.
En la literatura existen casos aislados tratados con otras modalidades terapéuticas, como la crema de tretinoína al 0,1% para tratamiento de áreas extensamente afectadas36 o el ingenol mebutato en lesiones numerosas que no han respondido a otras terapias71.
Tratamiento basado en la patogenia molecularVismodegib (GDC-0449) actúa sobre la vía SHH inhibiendo específicamente al receptor SMO. Ha sido aprobado por la Food and Drug Administration para el tratamiento de CBC metastásico o localmente avanzado no candidato a cirugía o radioterapia5,72. En 2009 se llevó a cabo el primer ensayo fase I (NCT00607724), en el que se evaluó la seguridad y farmacocinética del fármaco a las dosis de 150mg/día, 270mg/día y 540mg/día, y se estableció como la dosis más adecuada la de 150mg/día73. La aprobación de este vismodegib en 2012 se basó en un ensayo multicéntico fase II ERIVANCE/ NCT00833417, en el que se demostró la eficacia de vismodegib a dosis de 150mg/día74. En la actualización del ensayo ERIVANCE, vismodegib alcanzó una respuesta objetiva en el 48% de los pacientes con enfermedad localmente avanzada y en el 33% de los pacientes con CBC metastásico75. Se han llevado a cabo otros ensayos clínicos en CBC localmente avanzado y metastásico empleando una dosis de 150mg/día del fármaco (NCT01160250, STEVIE/NCT01367665, RegiSONIC/NCT 01604252)76–78. De entre todos ellos, se encontró una mayor tasa de respuesta global en el ensayo STEVIE, que reportó respuesta objetiva del 66,7% en los pacientes con CBC localmente avanzado y del 37,9% en CBC metastásico, con una mediana global de tiempo hasta respuesta de 2,7 meses77. Los ensayos clínicos realizados hasta la fecha apuntan a que la eficacia y seguridad de vismodegib en los pacientes con SG que presentan CBC localmente avanzados y metastásicos es similar a la de los pacientes con CBC esporádico79. Así mismo, vismodegib ha demostrado producir una disminución en tamaño de los queratoquistes odontogénicos en pacientes con SG80.
Recientemente se ha publicado un nuevo ensayo clínico (MIKIE / NCT01815840), que evalúa dos regímenes de terapia intermitente con vismodegib en pacientes con múltiples CBC (más de 6 tumores), incluyendo pacientes con SG. Los pacientes se randomizaron al grupo de tratamiento A (vismodegib 150mg/día 12 semanas, seguido de 3 tandas seguidas compuestas por 8 semanas de placebo y 12 semanas de tratamiento con vismodegib 150mg/d) o B (vismodegib 150mg/día 24 semanas, seguido de 3 tandas seguidas compuestas por 8 semanas de placebo y 8 semanas de tratamiento con vismodegib 150mg/d). Ambas pautas consiguieron reducción en el número de CBC, aunque la disminución fue mayor con la modalidad A de tratamiento en el subgrupo de pacientes con CBC esporádico. Así mismo, la pauta A consiguió una mayor reducción del diámetro tumoral81.
Vismodegib se ha estudiado como tratamiento y profilaxis de CBC en pacientes con SG, empleándolo durante largos periodos de tiempo. Se ha llevado a cabo un ensayo clínico fase II doble ciego (NCT00957229), centrado en pacientes con SG con al menos 10 CBC. En estos enfermos, vismodegib redujo significativamente la incidencia de nuevos CBC operables comparado con placebo, disminuyó el tamaño de los tumores preexistentes significativamente y redujo el número de cirugías necesarias82,83. El principal inconveniente de los tratamientos a largo plazo son los efectos secundarios, que obligan a la suspensión del tratamiento en muchos pacientes y en consecuencia se produce recidiva tumoral. El 74% de los pacientes del ensayo NCT00957229 tuvo que interrumpir el tratamiento en algún momento debido a efectos adversos. La reintroducción del fármaco en SG no está asociada con pérdida de eficacia del mismo, pero también reaparecen los efectos secundarios83. Por este motivo, algunos autores proponen realizar tratamiento intermitente con vismodegib en SG para conseguir una mejor tolerancia a los efectos secundarios84.
Casi todos los pacientes en los ensayos clínicos presentaron efectos adversos, habitualmente de bajo grado, entre los que destacan calambres musculares, fatiga, disgeusia, anorexia, alopecia y pérdida de peso. Entre el 22 y el 32% de pacientes presentaron efectos adversos graves74,77.
Se ha observado que en algunos casos se crean resistencias a vismodegib, debido a mutaciones adquiridas en SMO85,86. Estas resistencias se adquieren de manera rápida y pueden comprometer el tratamiento85. Así mismo, se ha descrito también un aumento de queratoacantomas y carcinomas espinocelulares en pacientes tratados con este fármaco14,87 por activación secundaria de la vía RAS/MAPK al inhibir la vía SHH87.
Sonidegib (LDE225) es otro inhibidor de SMO aprobado por la FDA para pacientes con CBC localmente avanzado que no es susceptible a la cirugía curativa ni a radioterapia88. El principal estudio con respecto a este fármaco es el ensayo BOLT/ NCT01327053, en el que se testaron las dosis orales de 800mg/día y 200mg/día, obteniéndose una eficacia similar en ambas. Sin embargo, el perfil de efectos adversos más favorable para la dosis de 200mg/d hace que esta sea la dosis recomendada. La mayor parte de los pacientes experimentan también efectos secundarios. Los más frecuentes son mialgias, disgeusia, alopecia, náuseas, pérdida de peso y elevación de creatinkinasa89. Pueden crearse resistencias a sonidegib, y parece que los pacientes que desarrollan resistencias a vismodegib tampoco responden a sonidegib90. Además de la administración oral, sonidegib se ha empleado al 0,75% en crema en pacientes con SG con resultados prometedores91.
Saridegib (IPI-926) es un derivado de la ciclopamina, de administración oral, que antagoniza selectivamente a la proteína SMO. A dosis de 160mg/d, podría también inducir respuestas clínicas en pacientes con CBC, aunque el único ensayo clínico hasta la fecha recoge únicamente 39 pacientes con esta patología92.
Itraconazol es un inhibidor de SMO que impide la acumulación de esta proteína en el cilio primario93 y se ha ensayado a dosis de 100mg/12 h y 200mg/12 h, consiguiendo una reducción del área tumoral del 24%94. El arsénico y sus derivados también bloquean la acumulación de Gli-2 en el cilio primario e impiden su activación95. Tanto itraconazol como el trióxido de arsénico, solos o en combinación, inhiben la vía SHH in vitro e impiden el crecimiento tumoral en meduloblastomas y CBC de ratones con SMO mutado resistente a otros inhibidores de esta proteína96.
ConclusionesEl SG es una enfermedad de herencia autosómica dominante debida a mutaciones en genes de la vía SHH, fundamentalmente en PTCH1. Se caracteriza por la aparición de múltiples CBC junto con otras manifestaciones cutáneas y extracutáneas, por lo que es necesario un equipo multidisciplinar en el diagnóstico, seguimiento y tratamiento de estos pacientes. Las modalidades de tratamiento de los CBC asociados a esta enfermedad son múltiples, entre los que destacan la cirugía convencional, la cirugía micrográfica de Mohs, la TFD e imiquimod al 5% tópico. Los estudios recientes en fármacos inhibidores de SHH como vismodegib están permitiendo un nuevo enfoque terapéutico en estos pacientes, aunque en muchos casos los efectos secundarios y las resistencias condicionan su eficacia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Queremos agradecer a las Dras. Ángela Hernández Martín y Elena García Esparza su ayuda en la elaboración de este artículo, aportando fotografías de sus pacientes.


