



A 48-year-old woman with no previous history of interest was assessed in the dermatology department for a lesion on the mons pubis that had first appeared several months earlier. The lesion had not responded to intralesional corticosteroids. The patient reported no scarring or previous folliculitis.
Physical ExaminationPhysical examination revealed a painless indurated multinodular plaque with a smooth surface and well-defined borders measuring 4cm in diameter on the mons pubis (Fig. 1). The patient also had 2 erythematous papular lesions measuring less than 1cm on both sides of the upper chest. She had no systemic symptoms or enlarged lymph nodes.
Histopathology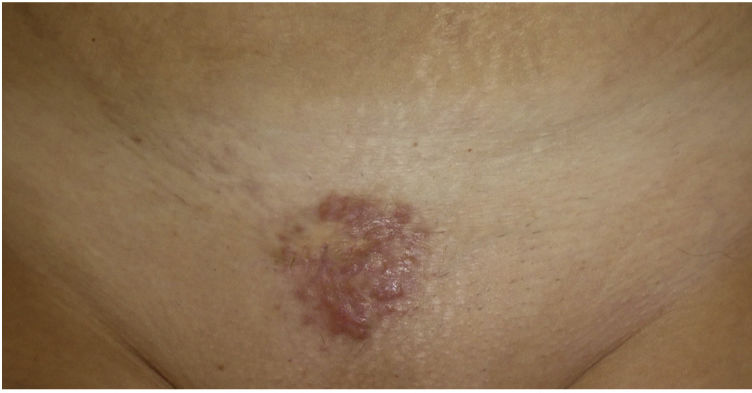
Biopsy revealed a normal epidermis and a dense polymorphous infiltrate in the middle and deep dermis, as well as increased collagen fibrils and vascularization (Fig. 2). There was evidence of emperipolesis.

Immunohistochemistry revealed positive findings for CD68, S100 (Fig. 3), and cyclin D1 and negative findings for CD1a.
Additional Tests
An extension study was performed with computed tomography and a full laboratory workup, neither of which revealed abnormalities. Similarly, serology testing was negative for Epstein-Barr virus, cytomegalovirus, human herpesvirus (HHV) 6, HHV8, HIV, and syphilis, and skin cultures were negative for Mycobacterium and Treponema species.
What is your diagnosis?
DiagnosisCutaneous Rosai-Dorfman disease.
Clinical Course and TreatmentThe lesions were surgically removed. A new lesion appeared on the patient's upper chest after 4 months of follow-up. This was removed, and the same histology findings were reported.
CommentRosai-Dorfman disease is a proliferative non-Langerhans cell histiocytic disorder that generally progresses with the presence of massive painless bilateral enlarged lymph nodes, systemic symptoms, increased acute phase reactant values, and hypergammaglobulinemia. Extranodal manifestations are observed in up to 40% of patients, with the skin affected in approximately 10%.1
One form of Rosai-Dorfman disease only affects the skin. Mean age at onset is higher than that of the classic form (43.5 vs. 20.6 years), and the disease predominantly affects women.1
The most common clinical presentation is the papulonodular form, although a wide variety of lesions have been reported (indurated plaques, tumors, acneiform lesions, xanthogranulomatous lesions, and ulcerations). Consequently, the differential diagnosis is very varied and includes histiocytic, inflammatory, and tumor-like proliferations (e.g., cutaneous lymphoma, Kaposi sarcoma, dermatofibrosarcoma protuberans), as well as skin infections caused by mycobacteria. The disease mainly affects the thorax, and there is frequently more than 1 lesion at diagnosis.1
Rosai-Dorfman disease has been associated with viral infections, especially those caused by herpesvirus, and mutations in MAP2K1/ERK, proteins that form part of one of the pathways that regulate cell survival and proliferation. An association has also been established with autoimmune diseases, such as systemic lupus erythematosus and juvenile idiopathic arthritis, as well as with lymphoid and myeloid neoplasms. A similar form has been described with mutations in SLC29A3 in the germline. This gene has been involved in H syndrome and in pigmented hypertrichosis with insulin-dependent diabetes mellitus. Lastly, high levels of IgG4 have been detected, especially in the classic form with visceral involvement, thus opening the debate on whether this condition should be included within IgG4-related diseases.2
Diagnosis is based on histopathology of the lesion and on a set of additional tests to rule disease at other levels. These include imaging tests, mainly computed tomography, and a laboratory workup comprising a complete blood count, biochemistry, kidney and liver function, antinuclear antibodies, complement, proteinogram, and serology testing for Epstein-Barr virus, cytomegalovirus, HHV6, HHV8, HIV, and syphilis.3
Biopsy reveals a dense histiocytic infiltrate in the dermis with lymphocytes, plasma cells, and disperse neutrophils. The presence of emperipolesis is a key diagnostic finding. Immunohistochemistry shows the origin of the cell strain that makes up the lesion and is positive for CD14, S100, CD68, and CD163 (histocyte markers) and negative for CD1a, langerin (Langerhans cell markers), and factor XIIIa.1
The lesion can be treated with surgery,4 as well as with oral and intralesional corticosteroids, methotrexate,5 topical imiquimod,6 and cryotherapy. Isolated cases treated using these approaches have been reported. Prognosis is generally good, potentially with spontaneous remission of the lesions in up to 20% of cases.4
Conflicts of InterestThe authors declare that they have no conflicts of interest.


