








Paraneoplastic pemphigus (PNP), a subset of pemphigus, is a unique autoimmune blistering condition that can affect multiple organs other than the skin. It is a life-threatening disease associated with an underlying malignancy, most commonly of lymphoproliferative origin. The clinical picture may resemble pemphigus, pemphigoid, erythema multiforme, graft-versus-host disease, or lichen planus. The earliest and most consistent finding is a painful, severe, chronic and often recalcitrant stomatitis. Treatment of PNP is difficult. Immunosuppressive agents are required to decrease blistering, and treating the underlying tumor may control autoantibody production. In this review, we included essential diagnostic aspects of PNP and the most useful treatment options in the dermatologist practice.
El pénfigo paraneoplásico (PNP), una variedad de pénfigo, es una enfermedad ampollosa autoinmune que puede afectar a múltiples órganos distintos de la piel. Es una enfermedad grave asociada con una malignidad subyacente, comúnmente de origen linfoproliferativo. Las lesiones clínicas pueden parecerse al pénfigo, penfigoide, eritema multiforme, enfermedad de injerto contra huésped o liquen plano. El hallazgo más temprano y más consistente es una estomatitis dolorosa, grave, crónica y, a menudo, recalcitrante. El tratamiento del PNP es difícil. Se requieren agentes inmunosupresores para disminuir la formación de ampollas y el tratamiento del tumor subyacente puede controlar la producción de autoanticuerpos. En esta revisión se incluyeron los aspectos diagnósticos más esenciales del PNP y las opciones de tratamiento más útiles en la práctica dermatológica.
In 1990, Anhalt et al.1 reported a new clinical entity named as “Paraneoplastic pemphigus”, fulfilling five diagnostic criteria. Subsequently, their findings were confirmed by several studies.2–9 Nguyen et al.10 described PNP as a heterogeneous autoimmune syndrome that affects several internal organs, and that its pathophysiology is not limited to antibodies targeting adhesion molecules like other subtypes of pemphigus. In 2001, they proposed the term “Paraneoplastic autoimmune multi-organ syndrome” instead of PNP. This is because the autoantibodies in the disease bind to the kidney, smooth and striated muscle, as well as the epithelium of the small intestine, colon and thyroid.3 The term more widely used in most reports and reviews, including the present paper, is PNP. About the position of PNP among skin disorders, some studies11–13 propose to include it as a type of pemphigus with an associated tumor, whereas others14–16 describe it as an independent autoimmune disorder; moreover, PNP does not fully meet Curth's criteria for cutaneous paraneoplastic syndrome (Table 1).
Curth's criteria for the diagnosis of cutaneous paraneoplastic syndrome.
| Criteria |
|---|
| Both conditions began simultaneously (neoplasia and paraneoplasia). |
| Development of a parallel course (treatment of the neoplasia results in regression of the skin lesion; recurrence of the neoplasia implies recurrence of the skin lesion). |
| The skin lesion is not associated with a genetic syndrome. |
| There is a specific type of neoplasia that occurs with paraneoplasia. |
| The dermatosis is rare in the general population. |
| There is a high frequency of association between both conditions. |
PNP: paraneoplastic pemphigus.
Paraneoplastic pemphigus is closely related to benign or malignant tumors. The most often reported malignancies are lymphomatoid and hematologic (B-cell lymphoma, chronic lymphocytic leukemia, Castleman's disease, Waldenstrom's macroglobulinemia, and thymoma, with or without myasthenia gravis). Interactions between the immune system and concomitant neoplasm seem to be key pathogenic steps with autoantibodies directed against both desmosomal and hemidesmosomal antigens. In PNP, most patients develop autoantibodies against periplakins and envoplakins.
In 1990, Anhalt et al.1 first described five cases of patients with a rare form of atypical pemphigus that were all associated with lymphoproliferative diseases. PNP mostly affects adults between 45 and 70 years old, but it may also be found in younger patients, in whom Castleman's disease is more commonly seen. There is no known correlation between incidence of the disease and specific gender, race, or geographical distribution.3
Based on its very unique clinical pictures, as well as its histologic and immunologic features, and most of all its elevated mortality (90% if untreated), diagnosis should be stated promptly.3,17–20 Prognosis depends on the nature of the associated tumor. Some patients experience rapid improvement after excision of a benign tumor, such as PNP associated to Castleman's disease. However, malignant tumors are often accompanied not only by higher mortality from the associated malignancy but also because the PNP can be severe and often recalcitrant.
EpidemiologyThe exact incidence of PNP is unknown but it is less common than pemphigus vulgaris or pemphigus foliaceus. There appears to be no age preference.4 Although PNP presents most often in older patients aged between 45 and 70 years, it also occurs in younger patients. The disease has been reported in patients ranging from 7 to 83 years-old.5 Ogawa et al.21 studied 496 patients with malignancy and recorded 25 cases of pemphigus (5%), an elevated number when compared with controls. There was a positive correlation with advancing age. The mean age of pemphigus patients with malignancy was 64.7 years.21 It appears to be no gender predilection.2,22
The associated malignant or benign neoplasm may be occult or already diagnosed at the point of PNP presentation. PNP may also develop after the tumor has been treated.6,7 The most commonly associated tumors are hematological, accounting for nearly 84% of all cases, these include non-Hodgkin's lymphoma (38.6%), chronic lymphocytic leukemia (18.4%), Castleman's disease (18.4%), thymoma (5.5%), Waldenström macroglobulinemia (1.2%), Hodgkin's lymphoma (0.6%) and monoclonal gammopathy (0.6%).4 Non-hematological neoplasms include carcinomas (8.6%), sarcomas (6.2%)23 and melanoma (0.6%).4
In children and adolescents, PNP is most commonly associated with Castleman's disease8 and PNP is often the presenting sign of Castleman's disease.9 This tumor is rare in the general population but is the third most common neoplasm associated with PNP.24
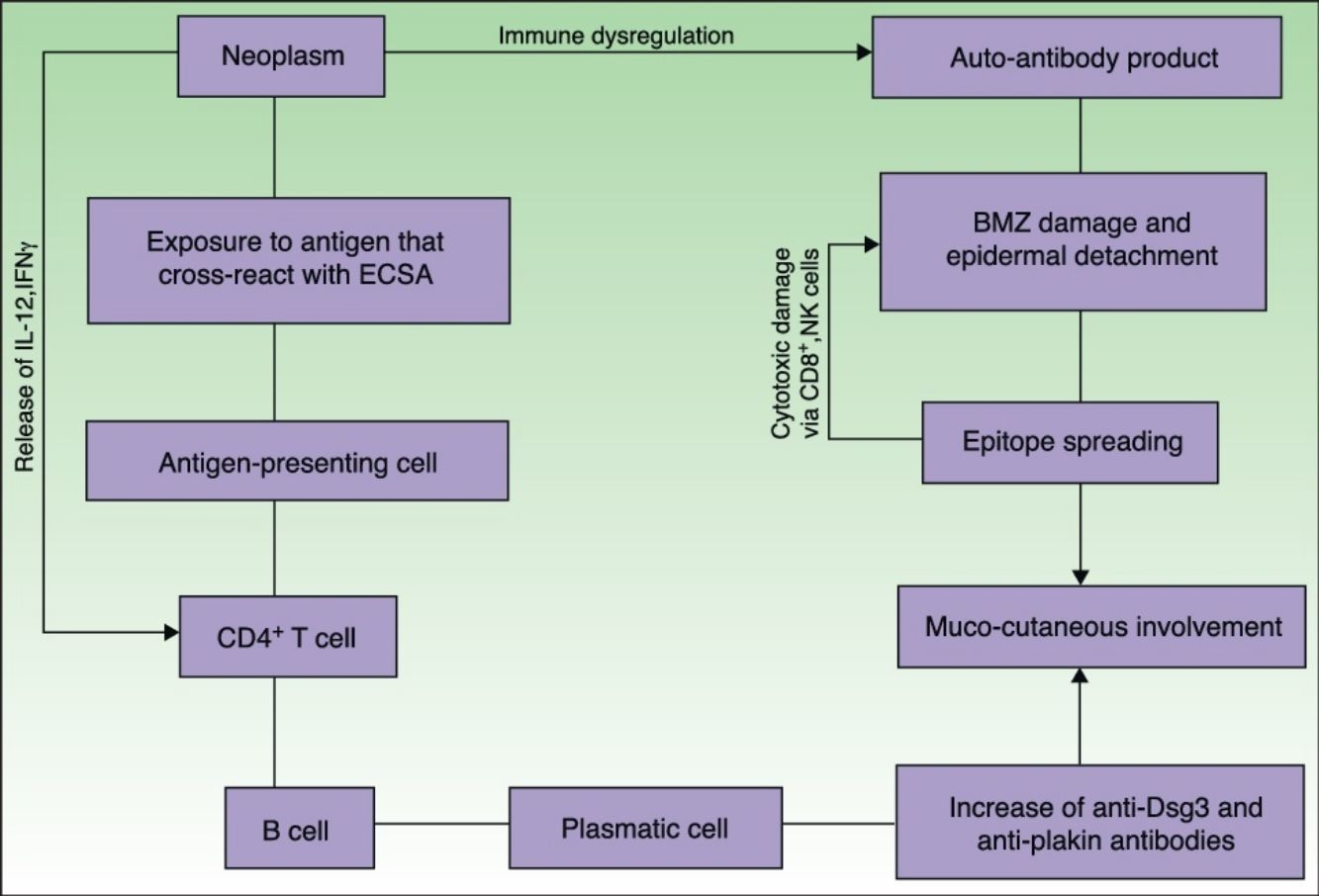
EtiopathogenesisPNP is an autoimmune disorder launched by an underlying neoplasm (Fig. 1). Etiopathogenesis of PNP is not fully described.4 Skin lesions are thought to be originated by an antibody-mediated autoimmune response to tumor antigens that cross-react with epithelial antigens. Tumor autoantibodies produce and release cytokines (such as interleukin-6) that enhance B-cells differentiation5 and foster to develop the humoral response.
. This mechanism can be enhanced by the epitope spreading creating secondary epitopes, increasing antibody production; this mechanism contributes to epidermal and subepidermal lesions (pemphigus and pemphigoid-like lesions). Cell-mediated immune activation (mainly CD8+ T cells) also develops and contributes to several skin lesions (lichenoid and graft-versus-host disease-like).")
Pathogenesis of paraneoplastic pemphigus. Neoplasms develop immune dysregulation and therefore the production of autoantibodies against self-antigens. Antigen-processing cells assimilate antigens that are cross-reactive with several skin antigens, presenting them to CD4+ T cells, and therefore to the known immune cascade that ends in the formation of autoantibodies against different substrates. The tumor itself can develop an immune process with the release of proinflammatory cytokines (IL-12 and IFN). This mechanism can be enhanced by the epitope spreading creating secondary epitopes, increasing antibody production; this mechanism contributes to epidermal and subepidermal lesions (pemphigus and pemphigoid-like lesions). Cell-mediated immune activation (mainly CD8+ T cells) also develops and contributes to several skin lesions (lichenoid and graft-versus-host disease-like).
PNP is often a clinical marker of benign and malignant neoplasms, most commonly malignancies of the lymphatic system.6 Ohzono et al.7 described the associated tumors in 104 PNP cases. Their clinical and histopathological findings were similar to those in previous reports.3,4,6
Some patients have tumors that are difficult to diagnose, such as follicular dendritic cell sarcomas located in the retroperitoneal space.8 Studies of patients with non-Hodgkin lymphoma revealed that most severe lesions during the PNP occur 2–3 years after diagnosis of lymphoma.6 Castleman's disease, also known as giant lymph node hyperplasia, occurs most commonly in children.
Previous studies have shown that HLA-DR4 and DR14 alleles confer strong susceptibility to pemphigus vulgaris and foliaceous; however, PNP is not associated with these alleles. HLA-DRB1*03 in Caucasian patients24 and Cw*14 in Chinese patients have been reported in PNP.25 These findings indicate that people with different racial backgrounds have a different susceptibility to PNP.
It is hypothesized that tumor antigens evoke not only a humoral response but a cellular one.4,5 While direct immunofluorescence findings of autoantibodies bound to the cell surface of affected epithelium support a humoral response, histopathological findings of individual keratinocyte necrosis with lymphocyte exocytosis support the role of cell-mediated immunity.
In order to explain such hypothesis, several explanations have been proposed, like production of autoantibodies to epithelial proteins by tumors, supported by finding B-cells-producing IgG antibodies directed to epidermal antigens in Castleman's disease26; cross-reactivity of tumor antigens and epidermal antigens; high levels of interleukin-6, which promotes B-cell differentiation and immunoglobulin production.27 Anti-IL-6 monoclonal antibody inhibitors (Tocilizumab) have been successfully used for treating Castleman's disease28–30; epitope spreading and cellular immunity-mediated processes have been reported in previous studies.31,32 Cummins et al.32 reported a series of four patients with a lichenoid variant of PNP without detectable autoantibodies. They inferred that these patients had disease mainly mediated by cytotoxic T lymphocytes rather than autoantibodies. Nguyen et al.10 also showed the presence of cytotoxic T lymphocytes, macrophages, and natural killer cells in tissues affected by PNP. These theories support that both cellular and humoral immunity are implicated in the pathogenesis of PNP.33
ClinicalVarious lesions may occur in patients with PNP. Although characterized by severe oral mucositis (Fig. 2), a generalized polymorphous cutaneous eruption and pulmonary involvement may develop. Typically, the first symptoms are usually florid, painful as well as intractable stomatitis, and also, it can involve the vermilion of the lips, oro and nasopharynx, the nose (Fig. 2), tongue, esophagus, stomach; duodenum, intestines and the pulmonary epithelium, as well as the conjunctiva34 and anogenital region (Fig. 3) are also affected.2,3,35–38


The lesions are polymorphic, and symptoms include blisters, erosions, spots, papules, and plaques. Nikolsky sign can be present. Cutaneous lesions usually appear after the onset of mucosal lesions and may involve any site, mostly the upper body. Nguyen et al.10 proposed a classification including several clinical presentations of PNP. It is believed that each category occurs with nearly equal incidence.
PNP cutaneous lesions can be classified into several groups according to the types of changes:
- -
Pemphigus-like: flaccid blisters, erosions, crust, and erythema.
- -
Bullous pemphigoid-like: scaly erythematous papules and stretched vesicles. These are more commonly seen on the extremities.
- -
Erythema multiforme-like: polymorphic changes, mainly erythematous peeling pellets with erosions and sometimes even with recalcitrant ulcerations (Fig. 4).
- -
Graft-versus-host disease: scattered dusky red scaly papules.
- -
Lichen planus-like: the presenting picture consists in flat, red-brown scaly papules and plaques, as well as intense mucous membrane involvement, more commonly seen in children on the trunk and extremities (Fig. 3), and rapidly extending to the face and neck.38 Scaly lesions on the palms and soles may accompany the lichenoid lesions.

Patients with PNP can develop life-threatening restrictive bronchiolitis obliterans. The frequency of the involvement of the respiratory system and its pathological mechanisms are unknown.39 In a study of 17 patients with PNP, restrictive bronchiolitis was found only in three patients. However, in another analysis where 28 patients with PNP and concomitant Castleman's disease were examined, the respiratory system was affected in 26 cases.40,41 Pulmonary disease, when present, is irreversible despite aggressive therapy.2,3,42 The recently discovered autoantigen, epiplakin, has demonstrated correlation with development of bronchiolitis obliterans in Japanese patients.43 Epiplakin is present in the respiratory bronchiole, and mice injected with epiplakin autoantibody showed abnormal changes in the histopathology of their pulmonary epithelia. While more research is needed, these early results indicate that epiplakin may represent a specific autoantigen in PNP-related bronchiolitis obliterans.
Differential diagnosis of PNP is extensive (Table 3), and includes pemphigus vulgaris, mucous membrane pemphigoid, erythema multiforme, Stevens–Johnson syndrome, lichen planus, graft-versus-host disease, and herpes simplex virus infection. When PNP is suspected, an extensive baseline workup should be conducted, including: blood cell count, lactate dehydrogenase, flow cytometry, as well as chest, abdomen, and pelvis CT scan. The existence of a neoplasm is often recognized prior to PNP (30% of cases approximately).22
HistologyThe disease often require several biopsies to achieve diagnosis.44 Horn and Anhalt45 examined 16 skin and oral mucous membrane biopsy specimens from six patients with PNP, and observed epidermal acantholysis, suprabasal cleft formation, dyskeratotic keratinocytes and vacuolar changes in the basal epidermis, and epidermal exocytosis of inflammatory cells. According to the morphology of the clinical lesions, the histopathology may reveal a different spectrum from minor inflammatory bullous lesions to a dense lichenoid reaction.45 Additionally, there might be vacuolar degeneration of the basal layer associated with band-like infiltrate of lymphocytes in the dermis (lichenoid features). Prompt clinic-pathologic correlation is recommended in these patients, as well as evaluations for neoplasm.
ImmunologyImmunopathology plays an important role in the diagnosis of PNP. The variety of possible autoantigens and the combination in which they occur, account for the diverse nature of this disorder, and thus for the conflicting findings.46–48 DIF performed on a perilesional biopsy may reveal intercellular deposits of IgG and C3 autoantibodies. In addition, linear deposits of IgG or C3 in the basement membrane zone, due to autoantibody binding to BPAG1-2, may be present. This helps distinguish PNP from other types of pemphigus, in which immunoglobulin deposits are found between keratinocytes but not on the basement membrane.3,5
Direct immunofluorescence can be negative in a few PNP patients, however, most cases exhibit positive DIF and thus, it is necessary for diagnosing PNP. False negatives on DIF are common in PNP for mucosal biopsies when necrotic tissue is predominant.5 Another reason is that a significant proportion of lesions are lichenoid, with predominant cellular immunity instead of humoral immunity.5,32
The autoantibody profile of PNP has been recently studied.46,47,49 Elegant studies50–53 have suggested that patients with PNP have autoantibodies against the plakin family (e.g. envoplakin and periplakin). Antibodies against desmoglein 1 and 3 (antigens for classic pemphigus), however, these antibodies may play a role in the initial stages of the development of PNP.54,55 The presence of autoantibodies to plakins is a characteristic feature of PNP. Envoplakin and periplakin antibody levels are most specific,56 followed by desmoplakin I and II.
Paraneoplastic pemphigus is the result of either a humoral or cell-mediated responses. This combined pathogenesis is clinically expressed in the varied pictures of PNP in contrast with those of pemphigus vulgaris. Auto-reactive cellular toxicity, mediated by cluster differentiating T lymphocytes (CD8+ cytotoxic T lymphocytes), CD56+ natural killer cells, and CD68+ macrophages, has also been implicated.56,50
Indirect immunofluorescence shows that IgG antibodies bind to the stratified epithelium in the esophagus and other tissues from monkeys. In contrast to pemphigus vulgaris and foliaceus, these antibodies also bind with the transitional and cylindrical epithelium of the urinary bladder, bronchi, small intestine, and colon, as well as, to a lesser extent, with myocardium and skeletal muscles and thyroid epithelium.50
In a previous study, Anhalt et al. showed that rat bladder immunofluorescence testing technique has a sensitivity of 75% and a specificity of 83% for diagnosing PNP. This can be explained because the transitional epithelium of the rat bladder contains desmoplakin but not envoplakin, periplakin and desmoglein,5 and not all patients with PNP have antibodies against all the antigens of the plakin complex.50,57 Plakin autoantibodies have been found in specific diseases: anti-desmoplakin antibodies in pemphigus vulgaris and erythema multiforme, anti-periplakin antibodies in pemphigus foliaceous and toxic epidermal necrolysis and rarely, anti-envoplakin antibodies in pemphigus foliaceous and vulgaris.56–58
Poot et al.,59 in a previous study, showed that indirect immunofluorescence with salt-split skin, showing a cytoplasmic staining of all layers of the epithelium, was a highly specific pattern of PNP, similarly as positive rat bladder indirect immunofluorescence.
Immunoprecipitation is the most sensitive and specific test for measuring anti-plakin antibodies in PNP.59 A positive immunoprecipitation test qualifies as a major criterion for the diagnosis of PNP,2 however, it has limited availability. Alternatives for the detection of plakin autoantibodies include immunoblotting and ELISA. Immunoblotting can be performed using an extract of cultured human keratinocytes to detect all desmosomal proteins: desmoglein 3 (130kDa), desmoplakin 1 (250kDa), BP230, desmoplakin 2 (210kDa), envoplakin (210kDa), plectin (>400kDa), periplakin (190kDa), epiplakin,43 and occasionally desmoglein 1 (160kDa). Immunoblotting and ELISA may also be performed using recombinant fragments of periplakin and envoplakin.60,61
DiagnosisEven when there is no consensus about the diagnostic criteria for PNP; the diagnosis is based on the criteria of Anhalt et al.1 mostly on clinical and histologic observations, direct immunofluorescence, indirect immunofluorescence, and immunoprecipitation tests (Table 2), furthermore, Camisa and Helm14 have classified these criteria into major and minor signs. Three major or two major and two minor signs are required to make a diagnosis of PNP. The most common laboratory finding is the immunoprecipitation pattern which is characteristic1,62 and may allow prompt diagnosis and active management of PNP.63,64
Characteristics of paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome.
| Characteristic | Features |
|---|---|
| Clinical | Painful mucosal erosions with a polymorphous skin eruption culminating in vesicles/bullae in the context of an occult/confirmed neoplasm. |
| Histopathology | Suprabasal acantholysis with clefting and suprabasal acantholysis resembling pemphigus vulgaris; interface dermatitis and exocytosis of inflammatory cells into the epidermis resembling lupus erythematosus; dyskeratosis (keratinocyte necrosis) with suprabasal acantholysis is a clue to PNP; may resemble changes seen in erythema multiforme or graft versus host disease; a lichenoid band may be seen along the dermal-epidermal junction resembling lichen planus. |
| DIF | Deposition of complement and IgG in intracellular epidermal spaces and in the basement membrane zone in linear granular lesions. |
| IIF | IIF of patient serum with rat bladder epithelia shows intercellular staining. Confirmation of autoantibodies to periplakin and/or envoplakin. |
| Neoplasm | Association with neoplasms (in order of frequency: non-Hodgkin lymphoma); chronic lymphocytic leukemia; Castleman disease; carcinoma; thymoma; sarcoma (liposarcoma, leiomyosarcoma, reticulum cell sarcoma, malignant nerve sheath tumor); Waldenström's macroglobulinemia; Hodgkin lymphoma, monoclonal gammopathy; melanoma. |
PNP: paraneoplastic pemphigus; DIF: direct immunofluorescence; IIF: indirect immunofluorescence.
Differential diagnosis of PNP.
| Oral mucositis due to chemotherapy and other causes of severe oral ulceration |
| Pemphigus (vulgaris, drug-induced) |
| Bullous pemphigoid and other autoimmune blistering disorders (including epidermolysis bullosa) |
| Mucous membrane pemphigoid (Cicatricial pemphigoid) |
| Drug eruptions |
| Oral (erosive) lichen planus |
| Graft versus host disease |
| Erythema multiforme |
| Stevens–Johnson syndrome and toxic epidermal necrolysis |
| Major aphthous stomatitis |
The first diagnostic criteria made by Anhalt et al.1 include (1) characteristic clinical appearance and histopathology and, (2) detection of tissue bound, circulating autoantibodies via direct immunofluorescence, indirect immunofluorescence and immunoprecipitation studies.
Anhalt further described other characteristics of PNP such as painful stomatitis, a polymorphous skin eruption with histological findings showing lichenoid or acantholytic changes, supportive immunofluorescence findings showing intercellular and basement membrane binding, serum antibodies that bind simple, columnar, and transitional epithelium, coexistence of lymphoproliferative disorders, and the presence of anti-dsg, desmoplakin I and II, envoplakin, periplakin, bullous pemphigoid antigen 1, and plectin antibodies.9
Extension work-upIn patients suspected of PNP, an extensive work-up for an underlying associated malignancy must be performed if no tumor has been diagnosed so far. This should include a chest, abdomen and pelvis computed tomography scan.65
TreatmentTo date, PNP treatment has been rather disappointing. It is vital to define and treat the associated neoplasm in PNP. In patients with an operable tumor, a surgical cure is often the best chance of inducing remission of PNP.66 The skin and mucosal eruptions cause severe morbidity and are often recalcitrant.65,66 The response to therapy is generally poor. Firstly, treatment is aimed at decreasing the production of autoantibodies.67–69 A better prognosis can be expected when the neoplasm is less aggressive (e.g. thymoma and Castleman's disease).66,70–75
Concurrent to a medical workup in patients without operable neoplasms, several non-surgical treatments have proven effective in reducing symptoms in these patients.76 Initially, glucocorticosteroid therapy should be added (prednisone (0.5–1.0mg/kg)).77 Cutaneous lesions tend to crust over and heal faster than mucosal. Steroid-sparing agents can be added to glucocorticoid therapy to reduce the total steroid burden. The addition of steroid-sparing agents, such as azathioprine, cyclosporine A,77,78 and mycophenolate mofetil,74 may reduce steroid intake and thus limit potential side-effects; however, a high level of caution is advised in patients with PNP and confirmed malignancy, where immunosuppression is paramount and dictates the mainstay of treatment options. Stem cell ablation therapy using high-dose cyclophosphamide without skin cell rescue has been used in some cases,70,79 but this is hazardous.
Less conventional therapies may be considered when the first-choice therapy fails; when the patient is severely ill or when prompt intervention is required. While results with plasmapheresis and intravenous immunoglobulin have been disappointing,5 rituximab, a monoclonal antibody to CD20 on B-cells, has shown promising results in patients with underlying B-cell lymphoma.71,80 Patients can be treated with the lymphoma protocol at a dose of 375mg/m2 weekly for 4 weeks, or the rheumatologic protocol of 1g once and repeated in 2 weeks. Additional cycles may be administered every 6–12 months depending on clinical response and recovery of the B-cell (CD20-CD19) population. Rituximab is usually well tolerated, but notable adverse effects of treatment include infusion and allergic reactions. Severe, life-threatening anaphylactic reactions have occurred. For this reason, rituximab is infused in a monitored setting such as an infusion center where an allergy can be rapidly identified and treated.80 In addition, progressive multifocal leukoencephalopathy, a fatal and untreatable reactivation of Creutzfeldt Jakob virus in the brain, has been reported in association with rituximab.81 This reactivation occurs only in the setting of severe immunosuppression. Alemtuzumab, a humanized monoclonal antibody against CD52, has also been successfully when used to induce long-term remission in a patient with underlying B-cell chronic lymphocytic leukemia.75,82
The concomitant use of rituximab with intravenous immunoglobulin has proven successful in patients who do not respond to conventional therapy or using rituximab as monotherapy. Intravenous immunoglobulin at 2g/kg per monthly cycle is usually dosed. Intravenous immunoglobulin is well tolerated and it has shown to be effective, rapidly reducing pathogenic autoantibodies in patients with autoimmune bullous diseases. Another benefit of intravenous immunoglobulin is the fact that it can be added into the patient's existing treatment regimen without added concern of additional immunosuppression, becoming a popular approach among clinicians who treat PNP. Intravenous immunoglobulin's favorable safety profile makes it an obvious choice in patients who are often on complicated treatments for PNP and an underlying malignancy. However, the considerable high cost has limited its extensive use.77
Patients with PNP have an increased susceptibility to skin infections related to the loss of skin integrity and the use of potent immunosuppressant. Early treatment of secondary infections with proper systemic antimicrobial therapy is of significant relevance to prevent sepsis and death.83
Adequate analgesia should be provided as the lesions can be painful. Oropharyngeal ulcerations may also interfere with proper feeding, and a nasogastric tube may be required.77
Importantly, involvement of the respiratory tract epithelium can lead to bronchiolitis obliterans, respiratory insufficiency and subsequent death. A perioperative infusion of high-dose intravenous immunoglobulin during the surgical removal of Castleman's disease has been suggested to reduce the risk of bronchiolitis obliterans, which is the most common cause of death in PNP patients with Castleman's disease.61,65,66 Lung transplantation has been reported in managing progressive respiratory insufficiency caused by constrictive bronchiolitis in a 14 year-old patient with PNP and Castleman's disease.84
PrognosisThe overall prognosis of PNP is poor. The mortality rate ranges from 75% to 90%, being respiratory failure the main cause of death.81 The prognosis is better when the disease is associated with benign tumors and may even remit when tumors are excised.
The outcome of PNP, however, does not simply parallel the course of the underlying malignancy. Complications resulting from PNP and its treatment contribute significantly to morbidity and mortality. These include sepsis, multi-organ failure, gastrointestinal hemorrhage and respiratory failure related to bronchiolitis obliterans.83
Ohzono et al.7 in a study of 104 PNP patients, of whom 40 died; the main cause of death in these patients was bronchiolitis obliterans (n=16/40, 40%), while the second cause (n=24/40, 60%) was infection (mainly pneumonia) and the third cause related to the associated tumor.
Bronchiolitis obliterans correlates with poor prognosis. Pulmonary involvement occurs in 30–92.8% of patients and it is estimated that one-third of the deaths in PNP are due to pulmonary insufficiency. It is known that constrictive bronchiolitis can continue to worsen despite improvement in muco-cutaneous manifestations after immunosuppressive therapy and resection of tumor.85 A prompt diagnosis and early introduction of treatment are mandatory.
ConclusionsThrough this literature review, we acquired a global context of paraneoplastic pemphigus. We present the main clinical features, the fundamental principles of its etiopathogeny, review the diagnostic aspects that integrate criteria distinguishing the entity from others such as pemphigus vulgaris; the main associated tumors, where we share part of the experience obtained in these almost 20 years studying autoimmune bullous diseases; finally the main treatment options and the prognostic factors of the disease are discussed. We emphasize the importance of making a timely diagnosis and choosing the most appropriate treatment option for each specific case.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflict of interestsThe authors declare no conflict of interests.


