






Introducción
La osteoartropatía hipertrófica (OAH) es un síndrome caracterizado por acropaquia (dedos en palillo de tambor), periostosis y artritis. Se distinguen dos variedades de la misma, una primaria o idiopática, más conocida como paquidermoperiostosis (PDP) o síndrome de Touraine-Solente-Golé1, y otra forma secundaria, de mayor incidencia, consecutiva fundamentalmente a una enfermedad pulmonar o cardiaca a veces de carácter neoplásico2,3. La PDP viene definida por tres criterios mayores: paquidermia, periostosis y dedos en palillo de tambor. A esto se añaden varios criterios menores: seborrea con hiperplasia sebácea, foliculitis, acné, hiperhidrosis, cutis verticis gyrata, etc. La forma primaria se considera una enfermedad hereditaria, si bien sólo se encuentra historia familiar de la misma en el 25-38 % de los casos. Su incidencia aproximada es de un 5 % respecto a la forma secundaria (95 %). Su expresividad es variable, por lo que son infrecuentes las formas completas de este síndrome. Las manifestaciones cutáneas, aunque presentes, pueden pasar desapercibidas para el paciente y su entorno2-4.
A continuación se describe un paciente, remitido del Servicio de Reumatología, con una forma completa de OAH primaria o PDP.
Caso clínico
Un varón de 54 años, trabajador de la construcción. No tiene hijos. En sus antecedentes familiares destaca cosanguinidad de sus padres (parentesco de tercer grado), tiene un hermano sano. Entre sus antecedentes personales figura una fractura de tibia y peroné izquierdos en 1990. Había sido diagnosticado de enfermedad de Paget en 1992. Es bebedor habitual de 1 litro de vino diario.
Acudió al servicio de Reumatología por un dolor en los dedos de las manos, los pies y las rodillas, que se acentuaba al subir y bajar escaleras, y un dolor nocturno en las extremidades inferiores que mejoraba con antiinflamatorios no esteroideos.
Se le realizó un estudio radiológico (radiografías de cráneo, huesos largos, columna, pelvis, manos y pies) hallándose una hiperostosis en los huesos de los antebrazos, el fémur y la tibia con una periostitis acusada y lesiones de fractura consolidada en la tibia y el peroné izquierdos. En las radiografías de manos se observó una tumefacción de partes blandas, sobre todo en falanges distales, una periostitis y una hiperostosis de los huesos metacarpianos y primeras falanges (fig. 1). En las radiografías de ambos pies también se apreció una hiperostosis de huesos metatarsianos, osteofitos y una esclerosis en el pie derecho.
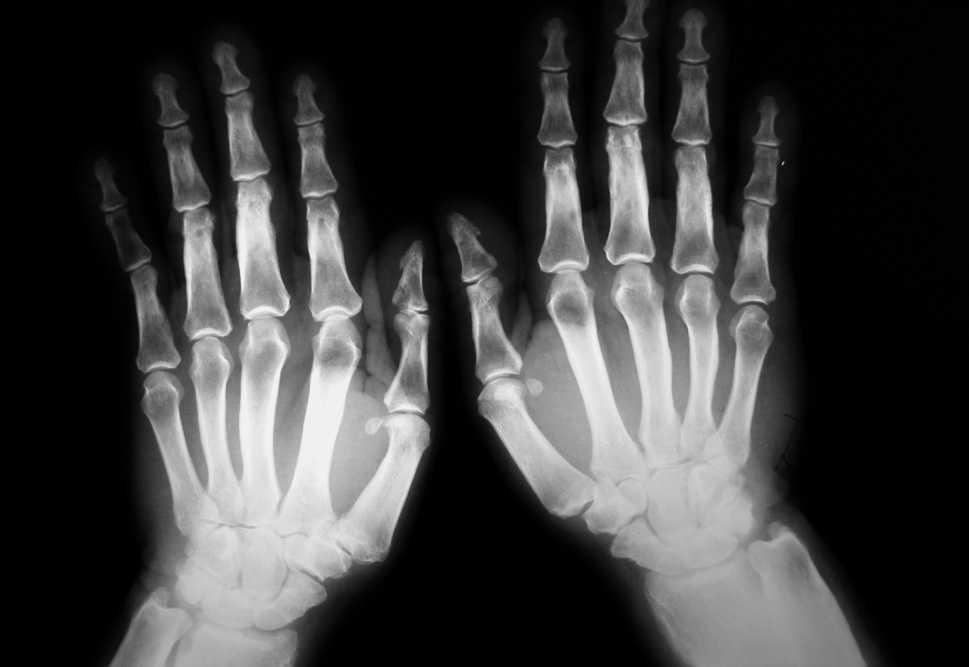
Figura 1. Hiperostosis y periostitis en huesos metacarpianos y primeras falanges. Tumefacción de partes blandas.
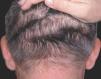
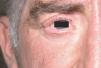
Las lesiones cutáneas, dado su desarrollo insidioso, habían pasado desapercibidas para el paciente y su entorno. Destacaba la presencia de pliegues muy pronunciados en la zona frontal, el entrecejo y los surcos nasogenianos (fig. 2). En la región occipital se dibujaban varios pliegues muy marcados (cutis verticis gyrata) (fig. 3). Los bordes palpebrales presentaban un engrosamiento (fig. 4). En conjunto, su cara tenía una expresión triste. La piel de las manos se mostraba tosca y los dedos voluminosos de aspecto acropáquico (dedos en palillo de tambor) (fig. 5). Los dedos de los pies tenían igual afectación, añadiéndose un engrosamiento ungueal. Los tobillos tenían apariencia edematosa.
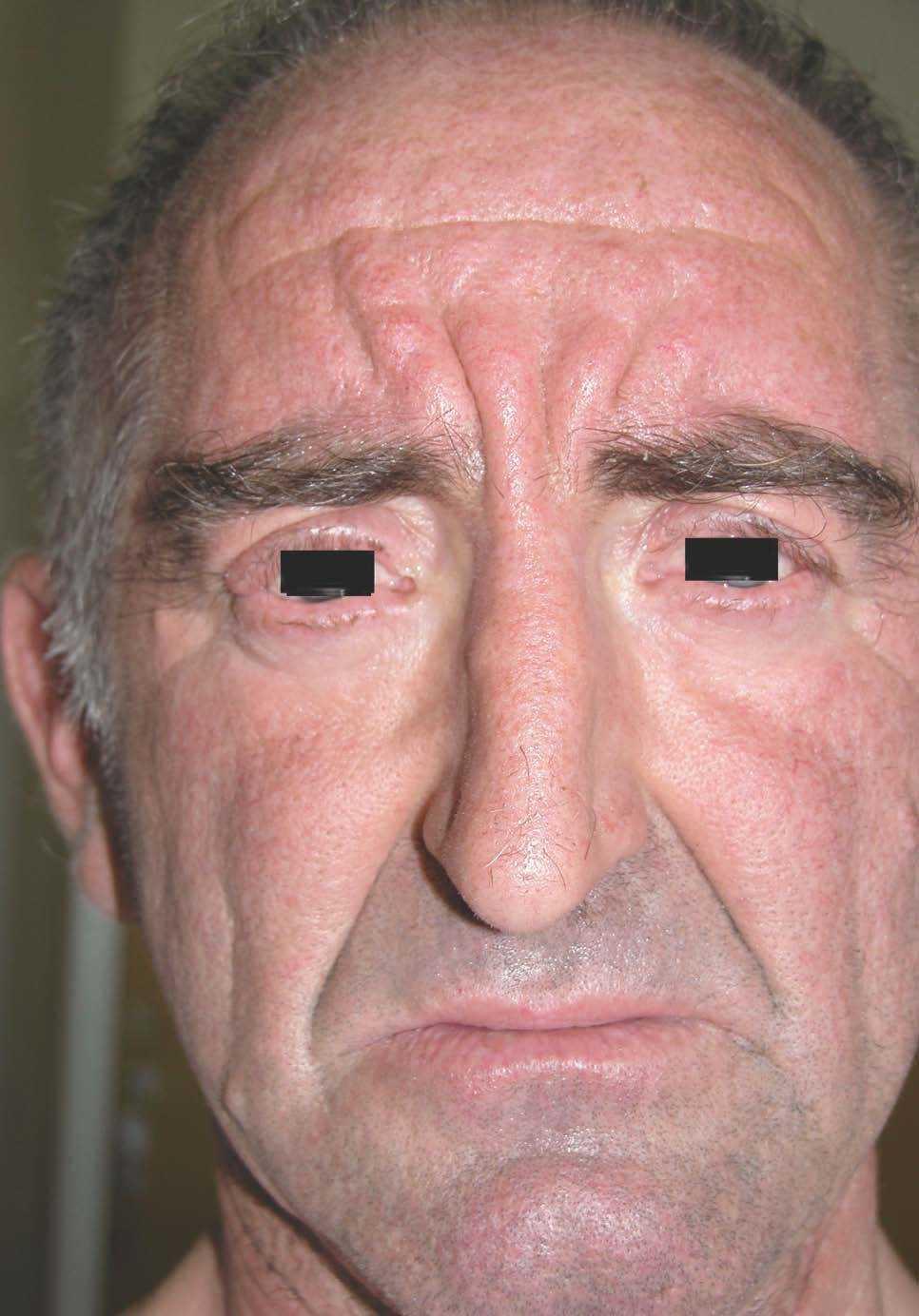
Figura 2. Pliegues cutáneos faciales muy marcados.
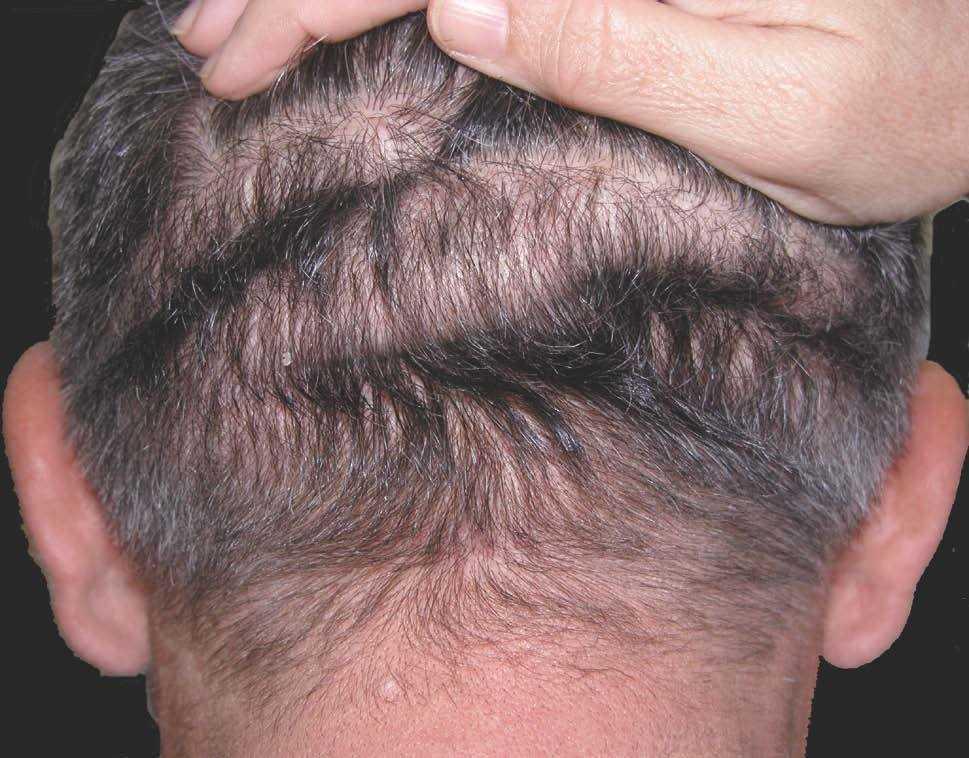
Figura 3. Pliegues occipitales (cutis verticis gyrata incipiente).
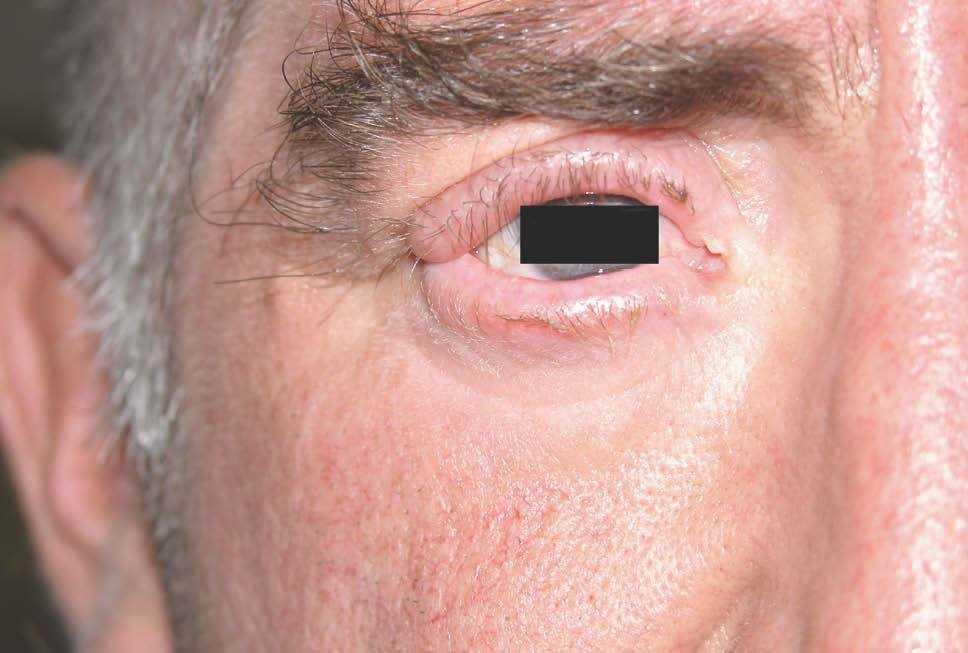
Figura 4. Borde libre palpebral superior e inferior engrosados.
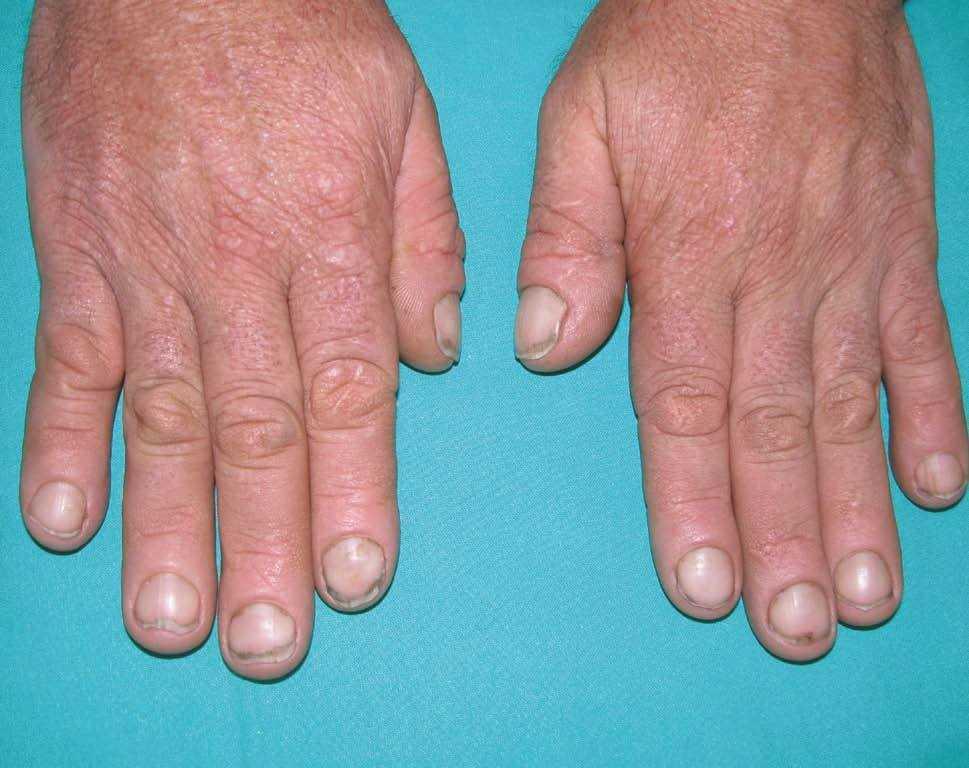
Figura 5. Manos con dedos acropáquicos.
Se realizó una analítica rutinaria que incluyó un hemograma, velocidad de sedimentación globular, ionograma, glucemia, uremia, creatinina, ácido úrico, calcio, fósforo, perfil hepático, lipídico y tiroideo, proteinograma, inmunoglobulinas, factor reumatoide y perfil urinario que no mostró alteraciones. La radiografía de tórax, el electrocardiograma y la gammagrafía ósea fueron normales.
La biopsia cutánea del dorso de una mano mostró una epidermis normal y un engrosamiento de la dermis a expensas de las fibras de colágeno (fig. 6).
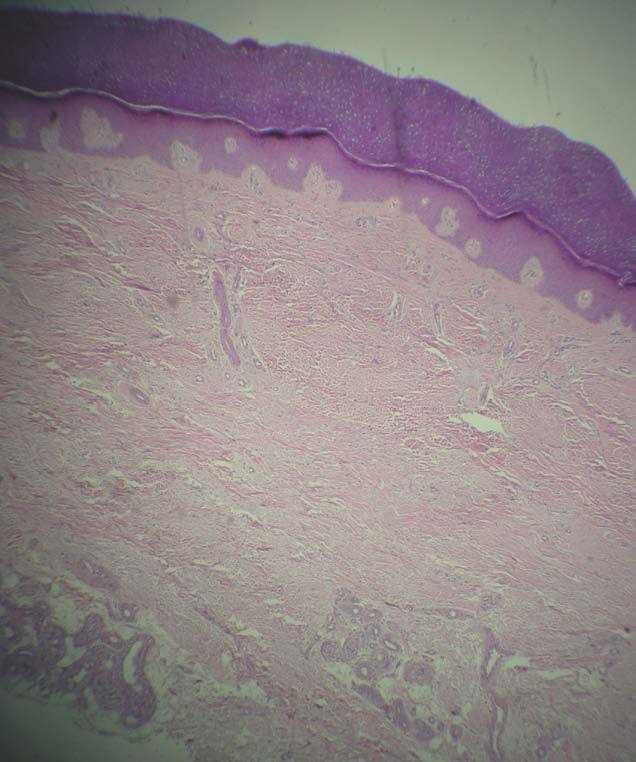
Figura 6. Engrosamiento difuso de la dermis a expensas de los haces de colágeno. (Hematoxilina-eosina, x60.)
Discusión
La OAH primaria también conocida como PDP es un proceso poco frecuente. Algunas de las alteraciones de este síndrome ya fueron descritas por Hipócrates 450 años antes de Cristo y también fueron observadas en esqueletos de América Central de la misma época5.
La primera descripción la realizó Friedrich en 1868 como «hiperostosis de todo el esqueleto»2,3. En 1890 Pierre Marie la definió como osteopatía hipertrófica pneumica2,3. Ya en 1935 fueron Touraine, Solente y Golé quienes individualizaron la PDP como forma primaria de osteopatía hipertrófica, denominándose también síndrome de Touraine-Solente-Golé1. Reconocieron que se trataba de una enfermedad familiar, con tres formas de presentación: una forma completa que cursaba con paquidermia y periostosis; una forma incompleta, sin la existencia de paquidermia, y una forma frustra o mínima, en la que existía paquidermia con mínimos cambios óseos.
La PDP predomina en varones en una proporción de 9:1. Es un proceso, a menudo familiar, transmitido de forma autosómica dominante con penetrancia incompleta, aunque también se han descrito casos transmitidos de forma autosómica recesiva y casos con antecedentes de consanguinidad como corresponde al nuestro, en que sus padres son cosanguíneos. También se han detectado anomalías cromosómicas y una mayor incidencia del HLA b12 en estos pacientes. Sus manifestaciones comienzan a observarse en la infancia y pubertad, progresan durante unos años y después se estabilizan. La paquidermia es el síntoma cutáneo más frecuente, el cual afecta a la cara y a las extremidades. Además, estos enfermos pueden presentar seborrea, acné, foliculitis, poros dilatados, hiperhidrosis de palmas, plantas y grandes pliegues y disminución del vello facial y pubiano4,6. Su aspecto clínico obliga a realizar diagnóstico diferencial con la acromegalia que también puede acompañarse de alteraciones cutáneas similares, incluso cutis verticis gyrata, pero los huesos de la cara, la mandíbula (prognatismo), el cráneo y las extremidades tienen un tamaño mayor en su conjunto y esto se hace muy evidente en el estudio radiológico en la ausencia de signos de periostosis4.
La clínica reumatológica, como sucedió en nuestro caso, suele ser el motivo de consulta debido a dolores óseos, derrame articular y artritis asimétrica3. Al realizar un estudio radiográfico se observa osificación irregular periostótica afectando sobre todo a los huesos largos, metacarpo, metatarso, falanges y epífisis. Además, puede apreciarse un engrosamiento de los tejidos blandos, calcificaciones de las inserciones musculotendinosas y, raramente, erosiones articulares7. Requiere realizar el diagnóstico diferencial con la artritis psoriásica y la artritis reumatoide8.
Otros síntomas, no cutáneos ni óseos, que pueden presentar estos enfermos son ginecomastia, retraso mental y anomalías periodontales4.
En la PDP se han descrito asociaciones a diversos procesos, algunos malignos: carcinoma epidermoide de la cara9, gastritis hipertrófica, úlcera péptica, adenocarcinoma gástrico10, enfermedad de Crohn, mielofibrosis11, etc. Pueden surgir complicaciones por aumento de tejidos blandos e hiperostosis como ptosis palpebral5, compresión de raíces nerviosas, alteraciones auditivas, cifosis, artrosis, osteonecrosis de la cabeza del fémur y síndrome del túnel del carpo4.
El interés de este cuadro también reside en la importancia de determinar si se trata de una forma primaria (PDP) o secundaria. La forma secundaria de OAH viene precedida por enfermedades pulmonares (tumores, abscesos, enfisema, bronquiectasias, fibrosis quística), cardiopatías (congénitas, endocarditis), procesos hepáticos (cirrosis, neoplasias), intestinales (neoplasias, enfermedad inflamatoria intestinal, poliposis), enfermedades tiroideas (enfermedad de Graves), etc. Las lesiones óseas, en esta forma secundaria, son de evolución más rápida y dolorosa, y los cambios cutáneos oscilan de leves a moderados. El pronóstico está ligado a la enfermedad de base6,12-14.
Ambas formas de osteopatía hipertrófica, tanto primaria (PDP) como secundaria, comparten la misma patogenia, la cual es poco conocida. Se baraja una teoría neurológica sugiriendo como factor etiológico la estimulación del arco neural vagal ya que el síndrome revierte, en algunos pacientes, después de la vagotomía15. Más recientemente, se han implicado alteraciones en la funcionalidad de los fibroblastos, con un incremento de la síntesis de fibras de colágeno8. También se ha apuntado al posible papel que pueden ejercer las plaquetas con sus potentes factores de crecimiento8,10. El consumo de alcohol empeora el proceso en algunos casos4. Se admite cierta inestabilidad cromosómica ya que, trastornos genéticos como el xeroderma pigmentoso, la ataxia-telangiectasia y la anemia de Fanconi, entre otros, se han descrito asociados a la PDP9. Por último, una mayor hipersensibilidad de las células a estímulos externos (agentes físicos y químicos) puede justificar la predisposición de estos pacientes a desarrollar distintos tipos de cáncer10.
Muchos de estos enfermos no son biopsiados. Habitualmente, los hallazgos anatomopatológicos son superponibles a los encontrados en nuestro paciente: epidermis normal o acantósica, engrosamiento difuso de la dermis a expensas de los haces de colágeno y aumento de mucopolisacáridos ácidos. En fases más avanzadas se puede apreciar un engrosamiento capilar, un aumento del colágeno pericapilar y una hipertrofia de glándulas sebáceas y ecrinas4.
El tratamiento es sintomático para atenuar los dolores óseos, con antiinflamatorios no esteroideos, pamidronato o colchicina; la isotretinoína oral se ha utilizado para disminuir las alteraciones cutáneas como seborrea o acné. En caso de ptosis palpebral puede ser necesario el tratamiento quirúrgico5.
Nuestro paciente es portador de una forma completa de PDP o síndrome de Touraine-Solente-Golé ya que presenta hiperostosis, dedos en palillo de tambor y paquidermia. Sus padres son cosanguíneos, pero no hay evidencia de que padezcan la enfermedad. El motivo de consulta fueron las manifestaciones articulares; la acropaquia en manos y pies se había ido desarrollando de forma progresiva y no era relevante para el paciente, así como tampoco lo eran sus rasgos faciales. Con las pruebas complementarias realizadas y teniendo en cuenta el lento desarrollo del proceso, hemos descartado patología sistémica que nos hiciera pensar que se trataba de una forma secundaria de osteoartropatía.
Concluimos que el diagnóstico de PDP u OAH primaria (síndrome de Touraine-Solente-Golé), requiere un alto grado de sospecha clínica, dado que muchos de estos pacientes son diagnosticados durante años de forma errónea de enfermedad de Paget o acromegalia. Suelen ser inicialmente valorados por traumatólogos o reumatólogos. En un tercio de los casos se halla una historia familiar por lo que conviene observar a los parientes cercanos. Las lesiones cutáneas, si no son muy evidentes, pueden pasar desapercibidas. Puesto que no siempre hay historia familiar ni expresividad completa clínica o radiológica, para llegar a su diagnóstico es necesario aunar criterios clínicos cutáneos (paquidermia, acropaquia) y radiológicos (periostosis) y descartar fundamentalmente trastornos cardiacos o pulmonares que justifiquen el desarrollo de formas secundarias. Por último, incidir en que estos pacientes pueden desarrollar a largo plazo malignidades y complicaciones por exceso de crecimiento de tejidos blandos y óseos, y que por tanto, requieren un seguimiento periódico.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Manuela Yuste Chaves.
Río Coa, 34.
37008 Salamanca. España.
Correo electrónico: manuelayuste@hotmail.com
Aceptado el 15 de septiembre de 2006.


