



Introducción
La incontinentia pigmenti (IP) es una genodermatosis infrecuente con carácter multisistémico. En la actualidad solamente se acepta la llamada clásicamente IP 2 o forma hereditaria. Los casos descritos previamente como IP 1 o forma esporádica corresponden más bien a mosaicismos pigmentarios, y no a verdaderos casos de IP tal y como se entiende en el momento actual1,2.
Presentamos tres casos de IP dos mujeres y un varón con presentación clínica y curso evolutivo diferentes.
Casos clínicos
Caso 1
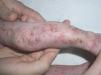
Una niña de 3 días de vida, nacida de embarazo y parto normales, sin antecedentes familiares de interés, es referida a nuestra consulta al tercer día de vida para valoración de lesiones vesiculosas interpretadas como herpéticas, tratadas sin éxito con aciclovir intravenoso. En la exploración física se objetivaron lesiones vesiculosas, erosivas, que alternaban con otras de aspecto costroso, siguiendo un trayecto lineal longitudinal en miembros inferiores (fig. 1), y trayectos lineales transversales y en «V» en tronco posterior. Más tarde las lesiones fueron evolucionando, y a las tres semanas de vida coexistían lesiones hiperqueratósicas junto con otras vesiculoerosivas en miembros inferiores, al mismo tiempo que aparecieron otras de aspecto verrucoso en porción acral de miembros superiores. Además, esta paciente desarrolló en un corto periodo de tiempo estrabismo y patología retiniana que desembocó finalmente en un desprendimiento de retina. El estudio anatomopatológico evidenció espongiosis eosinofílica y vesículas rellenas de eosinófilos, todo ello consistente con el diagnóstico de IP.
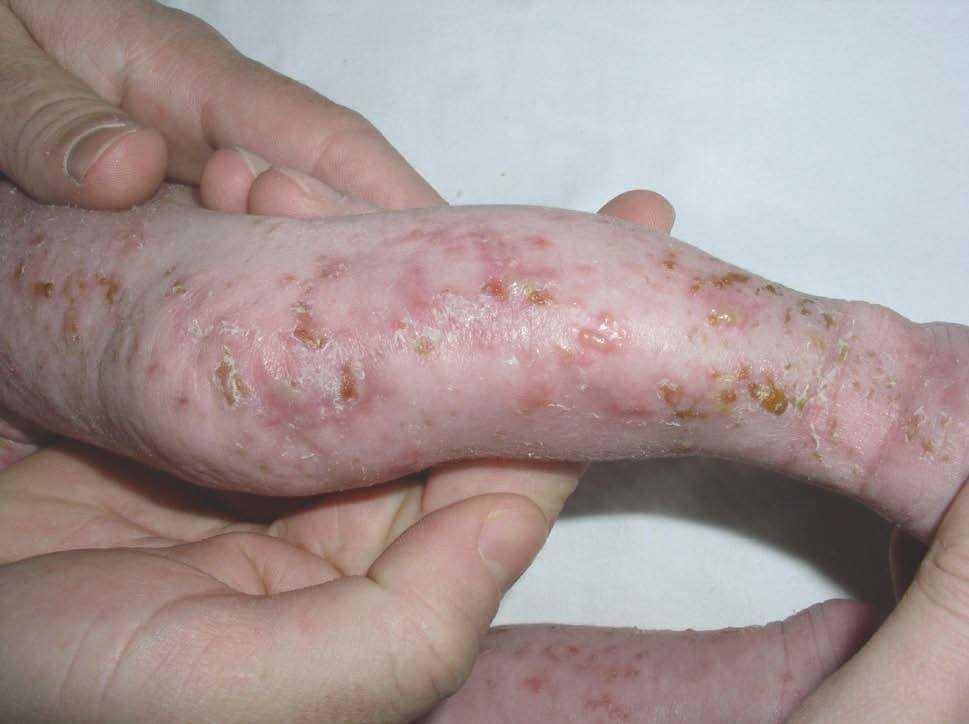
Figura 1. Eritema y vesiculación sobre base inflamatoria con distribución lineal longitudinal en extremidades inferiores (estadio 1). Placa verrucosa hiperqueratósica en rodilla (estadio 2).
Caso 2
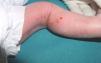
Una niña de 6 meses de vida es referida a nuestra consulta para valoración de máculas hiperpigmentadas que confluían en placas con disposición arremolinada en tronco anterior y en «V» en tronco posterior (fig. 2). En miembros superiores e inferiores tomaban disposición lineal, coexistiendo con placas hiperqueratósicas y lesiones verrucosas acrales. Según informes previos de su hospital de referencia, habían existido lesiones vesiculosas previas generalizadas, que habían remitido en el momento actual. Entre sus antecedentes familiares destacaban dos abortos maternos previos, ambos varones, en el quinto y sexto mes de embarazo. Además, la paciente presentaba clínica a otros dos niveles: oftalmológica, con retinopatía en estudio, y neurológica, con episodios convulsivos generalizados que se correlacionaron con malformaciones venosas vistas en resonancia magnética en territorio parietooccipital.
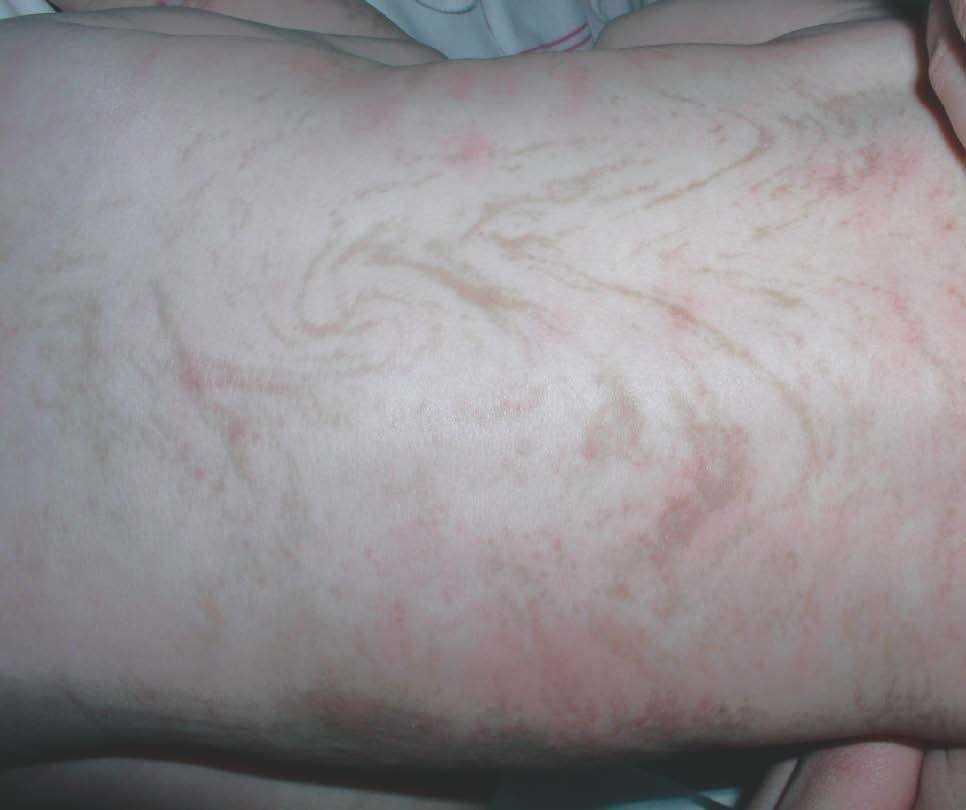
Figura 2. Máculas hiperpigmentadas con distribución blaschkoide en espalda (estadio 3).
Caso 3
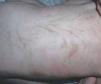
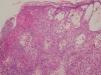
El último caso clínico corresponde a un varón, sin antecedentes familiares de interés, referido a nuestras consultas al sexto día de vida por lesiones papulovesiculosas lineales en miembro inferior izquierdo, que semanas después fueron desapareciendo, siendo sustituidas por otras lesiones hiperpigmentadas con descamación superficial más evidentes en fosa poplítea (fig. 3). Se planteó diagnóstico diferencial con otras entidades fundamentalmente NEVIL y se realizó estudio histológico, donde se evidenciaron espongiosis eosinofílica, vesículas rellenas de eosinófilos y disqueratosis focal (fig. 4), compatibles con el diagnóstico de IP. El cariotipo era normal, y tras tres años de seguimiento, permanece asintomático con involución espontánea de las lesiones cutáneas.
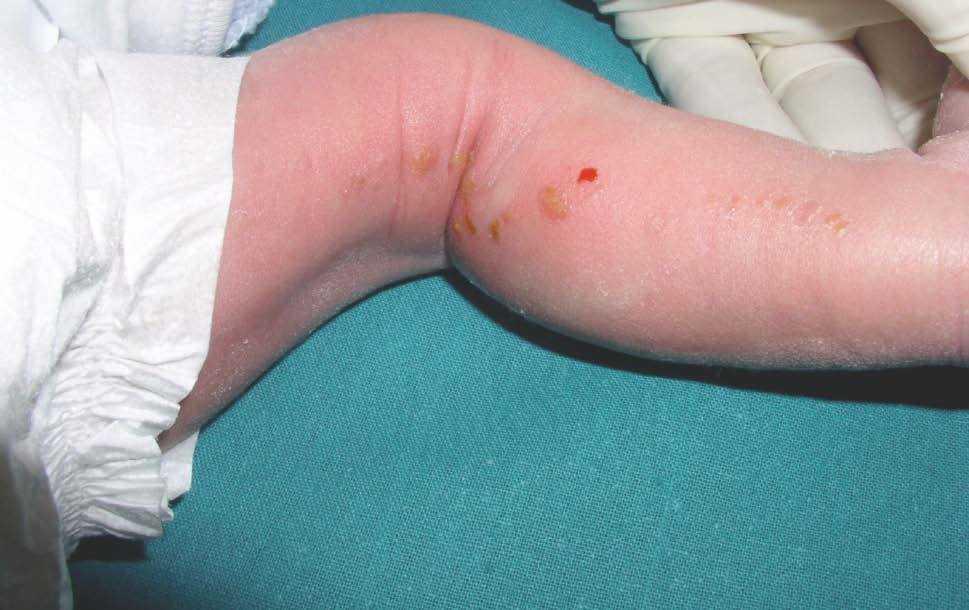
Figura 3. Eritema y lesiones papulovesiculosas lineales en miembro inferior izquierdo.
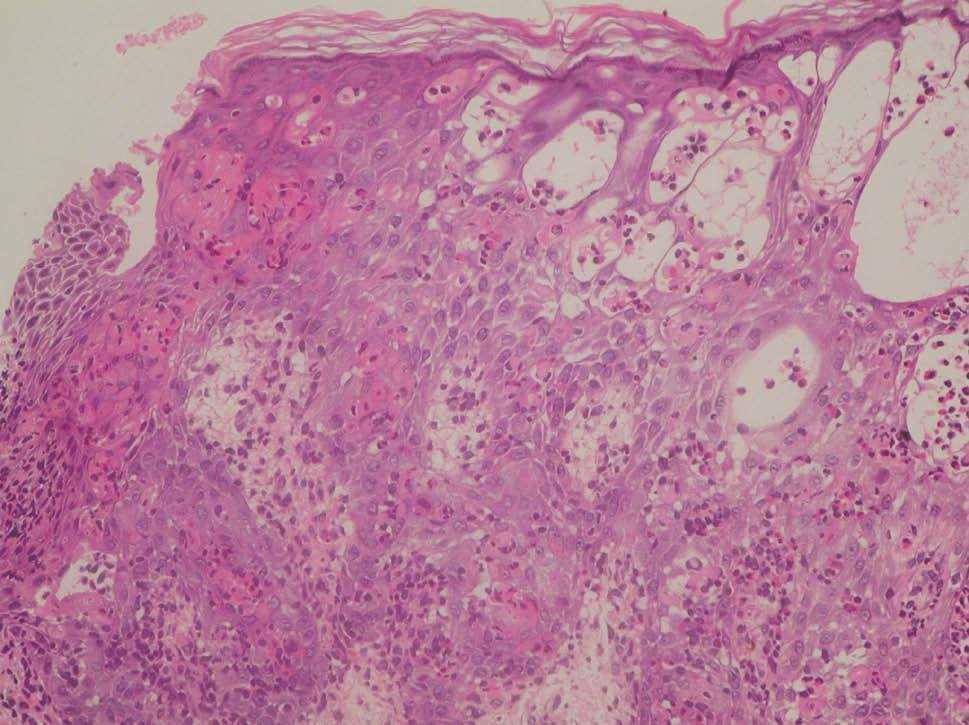
Figura 4. Dermatitis espongiótica eosinofílica, vesículas intraepidérmicas rellenas de eosinófilos y células disqueratósicas multiples; todos ellos son hallazgos histológicos típicos del estadio 1. (Hematoxilina-eosina, x20.)
Discusión
La IP es un trastorno multisistémico que se hereda de forma dominante ligado a X, lo que se demuestra por la mayor afectación de mujeres y la transmisión por líneas femeninas1-3. Al igual que en otras patologías con herencia ligada a X, la variabilidad clínica observada en mujeres es atribuida al fenómeno de lionización. La IP es letal en la mayoría de varones afectos intraútero, lo cual desemboca, en un gran número de casos, en aborto4. Recientemente se ha encontrado que las mutaciones localizadas en el gen NEMO (componente imprescindible en la vía de protección celular frente a la apoptosis inducida por la familia del factor TNF), localizado en el segmento Xq28, causan la enfermedad; es más, una única mutación provoca el 80 % de los casos, lo que tiene una consecuencia práctica inmediata: permite el consejo genético en un número importante de casos5. Los tres casos que hemos presentado previamente se encuentran pendientes en estos momentos del resultado del estudio genético.
Son excepcionales los casos de IP en varones, y por ello se recomienda la realización de un estudio genético completo, buscando alguno de estos tres mecanismos que faciliten la supervivencia de los mismos4: a) adición de un cromosoma X (por ejemplo, 47 XXY, que supondría un estado de heterocigosidad funcional); b) mosaicismo somático; c) alelos hipomórficos, mutaciones menos deletéreas en dominios diferentes del gen NEMO, que provocarían fenotipos diferentes, como ELA-ID6 (displasia anhidrótica ectodérmica más inmunodeficiencia) o ELA-ID-OL (lo anterior más osteopetrosis y linfedema), y que muchos autores sostienen que son un continuo con la IP. No se conoce bien la historia natural de la enfermedad en varones afectos supervivientes. En contra de lo que en un principio se podría esperar, y según la escasa literatura publicada hasta el momento, no tienen mayores tasas de mortalidad que las mujeres afectas, aunque harían falta más estudios y con mayor número de pacientes para aclarar este punto7.
La clínica típica de esta patología consta de cuatro estadios: vesiculoso (90 % casos), verrucoso (70 %), hiperpigmentado (98 %) y atrófico (42 %), como hemos presentado previamente1,2,8-10. Los diferentes estadios no son compartimentos estancos, sino que pueden solaparse entre sí; no obstante, las lesiones van cambiando de localización a medida que se pasa de un estadio a otro, teniendo predilección por el tronco y miembros en la fase vesiculosa y por los miembros, principalmente en zonas acrales, en la fase hiperqueratósica. De cara a un diagnóstico retrospectivo11,12, sobre todo en familiares de probandos, hemos de fijarnos en las lesiones de los dos últimos estadios: por un lado, las máculas hiperpigmentadas, que además de presentarse como grandes bandas con distribución blaschkoide, pueden hacerlo de una forma más sutil como máculas hiperpigmentadas de escasos centímetros de diámetro, más frecuentemente localizadas en región axilar, inguinal o mamaria. De igual forma las bandas atróficas, pálidas y sin vello, con predilección por la parte posterior de los miembros inferiores (propias del último estadio), pueden pasar inadvertidas si no se explora minuciosamente al paciente. Las series que incluyen más población adulta, principalmente a través de un diagnóstico retrospectivo, informan de mayor frecuencia de aparición de este último estadio y menor número de complicaciones neurooftalmológicas, por lo que se podría interpretar que otras series, de casos pediátricos fundamentalmente, pudieran sobreestimar las mismas al incluir solamente población pediátrica3.
Existen alteraciones extracutáneas a varios niveles; entre ellas destacan las dentarias, no sólo por ser las más frecuentes, sino porque al tener carácter permanente en muchas ocasiones colaborarán también al diagnóstico retrospectivo1-3,12. Pero es la clínica neurooftalmológica, como la de nuestros dos primeros casos, la que sin duda nos va a marcar el pronóstico13,14. La alteración oftalmológica cursa con visión normal en muchas ocasiones, de ahí que esté infradiagnosticada, pero de presentarse suele ser asimétrica y severa y asociada a patología neurológica. Las manifestaciones retinianas, junto con las neurológicas, suelen ser más tempranas, presentándose en el primer año de vida; sin embargo, las manifestaciones no retinianas son algo más tardías.
Con el fin de identificar tanto los casos típicos como los más sutiles Landy y Donnai en 1993 propusieron unos criterios diagnósticos, que pueden servir al clínico a modo de guía9 (tabla 1). En ellos se da mucho peso a los antecedentes familiares, de forma que cuando éstos están presentes los criterios menores pasan a ser mayores, y apoyan el diagnóstico de IP de igual manera que los clásicos criterios cutáneos. Por el contrario si no existen antecedentes familiares reconocibles, al menos un criterio mayor cutáneo es necesario para establecer el diagnóstico y unos criterios menores para apoyarlo. Los cambios histológicos de los diferentes estadios son característicos aunque no patognomónicos15 y desempeñan un papel importante en el diagnóstico, apoyando los hallazgos clínicos. Recurriremos al estudio genético, en líneas generales, para proporcionar consejo genético y para confirmar aquellos casos dudosos así como los que se presentan en varones.

En cuanto al tratamiento, no hay auténticos protocolos sobre el manejo de la IP, pero sí algunas guías que pueden ser de gran ayuda1. Así, la clínica cutánea cursa con resolución espontánea, y el tratamiento es sólo de apoyo. Es muy importante efectuar un diagnóstico y tratamiento precoz de la patología dentaria para evitar complicaciones secundarias, con controles radiológicos desde los dos años de vida, realizando cirugía correctora en caso necesario. Por otro lado, resulta imprescindible practicar una buena exploración neurológica en el momento del diagnóstico, recurriendo a las pruebas de imagen si se detectara alguna anomalía, tal y como se ha visto en el segundo caso clínico que hemos presentado anteriormente. Por último se recomiendan controles estrictos oftalmológicos14 con mayor periodicidad durante el primer año (tiempo en el que suele aparecer la patología retiniana) y posteriormente, hasta los tres o cuatro años, algo más espaciados.
Concluyendo, diremos que la IP es un trastorno infrecuente con herencia dominante ligada a X, por lo que la mayoría de casos comunicados corresponden a mujeres; los casos en varones son raros. Aunque es necesario un enfoque multidisciplinar para un correcto manejo de la misma, el diagnóstico recae principalmente en la clínica cutánea, ayudada de los estudios histológicos y genéticos. Queremos enfatizar la importancia de la clínica cutánea tardía (tercer y cuarto estadio) y de la clínica dental, ya que nos van a permitir en gran número de casos realizar un diagnóstico retrospectivo.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Marta Feito Rodríguez.
Servicio de Dermatología. Hospital Universitario La Paz.
Paseo de la Castellana, 261. 28046 Madrid. España.
Correo electrónico: marta8marta@hotmail.com
Aceptado el 15 de septiembre de 2006.


