





La papulosis linfomatoide es un proceso linfoproliferativo de células T CD30+ poco frecuente y de pronóstico excelente que generalmente afecta a adultos y, en menor medida a niños, por lo que tanto el espectro clínico-patológico como el riesgo de progresión a otro tipo de linfoma en el grupo pediátrico no está bien establecido.
ObjetivoAnalizar las características de la papulosis linfomatoide infantil a partir de la descripción de nuevos casos y de la revisión de la literatura.
Material y métodoSe realizó un estudio retrospectivo de 9 pacientes menores de 18 años diagnosticados de papulosis linfomatoide atendidos en nuestro servicio entre 1995 y 2009.
ResultadosSe incluyeron 7 niños y 2 niñas de edades entre 2 y 17 años. Las lesiones de papulosis linfomatoide se vieron precedidas en 2 casos y seguidas en 1 de otras compatibles con pitiriasis liquenoide aguda.
La resolución de las lesiones fue espontánea, dejando hiperpigmentación (77%) o hipopigmentación postinflamatoria (23%) y cicatrices en el 77% de los casos. Histológicamente todos los casos presentaron el patrón tipo A de papulosis linfomatoide. El estudio molecular mostró monoclonalidad en los 3 casos en los que fue realizado.
ConclusionesLa papulosis linfomatoide infantil es una entidad rara que se manifiesta clínicamente como la forma adulta. Esta enfermedad linfoproliferativa, que ocasionalmente se asocia a pitiriasis liquenoide aguda, muestra hallazgos histológicos compatibles con el patrón histiocitoide o tipo A. El desarrollo de otros procesos linfoproliferativos malignos en el seguimiento posterior es menos frecuente en la papulosis linfomatoide infantil comparado con la variante adulta. La frecuente asociación de pitiriasis liquenoide y de papulosis linfomatoide encontrada en nuestro análisis, así como la dificultad que supone en algunos casos el diferenciar entre ambos procesos, permite sugerir que ambas patologías podrían formar parte de un espectro clínico-patológico común.
Lymphomatoid papulosis is a rare lymphoproliferative T cell CD30+ disease with excellent prognosis which affects almost exclusively adult patients, being rarely in the childhood; thus the clinic and pathologic spectrum and the risk of evolution to another type of lymphoma are not well defined in the pediatric group.
ObjectiveThe aim of this article is to analyze the characteristics of infantile lymphomatoid papulosis and review the literature.
Material and methodA retrospective study analyzing the characteristics of 9 patients aged up to 18 diagnosed of lymphomatoid papulosis attended in our department from 1995 to 2009 was performed.
ResultsThe study included 7 boys and 2 girls aged between 2 and 17. Pityriasis lichenoides acuta's lesions appeared associated before and after lymphomatoid papulosis′ development in 2 and 1 cases respectively.
The lesions resolved spontaneously, leaving a postinflammatory hyperpigmentation (77%) or hypopigmentation (23%). The development of varioliform scars occurred in over 77% of cases. Histologically, all cases showed features compatible with type A of lymphomatoid papulosis. Molecular studies showed monoclonality in the 3 cases in which this technique was done.
ConclusionsInfantile lymphomatoid papulosis is a rare entity clinically manifested as the adult form. This lymphoproliferative disease, which is occasionally associated with pityriasis lichenoides acuta, shows features compatible with the type A or histiocytoid pattern in the histological analysis. The development of other lymphoproliferative disorders is less frequent in the infantile form than in the adulthood. The prevalent association among pityriasis lichenoides and lymphomatoid papulosis observed in our analysis, as well as the difficulties which supposed to differentiate between these two pathologies in various cases, suggest that those entities could be part of a common clinical and pathological spectrum.
La papulosis linfomatoide (PL) fue descrita por Macaulay en 19681,2 como una condición clínica benigna que asociaba características histológicas de un linfoma maligno. Previamente considerado como un proceso inflamatorio, actualmente se define como un linfoma cutáneo de curso indolente. Se trata de un cuadro clinicopatológico de etiología desconocida propio de la edad adulta que asocia hasta en un 20% de los casos un proceso linfoproliferativo maligno concomitante o en su seguimiento a largo plazo.
El desarrollo de la PL en la infancia es excepcional y sus características clinicopatológicas así como el riesgo de desarrollo de procesos linfoproliferativos malignos no están bien establecidas. Describimos 9 casos de PL en pacientes de edades comprendidas entre 2 y 17 años de edad y analizamos las características principales de este proceso en la edad infantil a partir de la revisión de los casos previamente publicados, identificando la existencia o no de diferencias respecto a la variante adulta.
Material y métodoPacientesSe realizó un estudio retrospectivo mediante la revisión de la historia clínica de todos aquellos pacientes con el diagnóstico confirmado de PL con edades comprendidas entre 1 y 18 años de edad, procedentes del Hospital infantil Niño Jesús, Madrid, España, tratados en el período comprendido entre el 1 de enero de 1995 y el 30 de septiembre de 2009.
Se analizaron los siguientes parámetros de cada uno de los pacientes: edad, sexo, lesión elemental y patrón de distribución del cuadro, número máximo de lesiones en un brote (1–5, 6–10, 11–20, mayor de 20) existencia o no de síntomas asociados, lesiones residuales, número de brotes al año, seguimiento a largo plazo (5 años de revisiones periódicas).
Análisis histológicoPara el estudio mediante microscopía convencional, las biopsias de los diferentes casos fueron fijadas con formalina al 4% e incluidas en parafina, para posteriormente ser teñidas con hematoxilina-eosina. El estudio inmunohistoquímico se realizó utilizando un inmunoteñidor automático (Dako Autostainer Link 48, Dako, Denmark) en combinación con el sistema de detección EnVision FLEX® (Dako, Denmark). Se utilizaron los siguientes anticuerpos: antígeno epitelial de membrana (EMA), CD2, CD3, CD4, CD8, CD20, CD79a, CD15, CD30 y CD68. El análisis molecular de reordenamiento del receptor de células T (RCT) fue realizado en 3 casos.
Los casos analizados se clasificaron morfológicamente según una reciente clasificación que diferencia 3 tipos de PL bien definidos: a) PL tipo A o histiocitoide, que se caracteriza por un infiltrado mixto compuesto mayoritariamente por linfocitos de pequeño tamaño junto a múltiples linfocitos atípicos de gran tamaño, acompañado de un número variable de neutrófilos y macrófagos, con algún eosinófilo aislado; b) PL tipo B o «tipo micosis fungoide», que se caracteriza por un infiltrado monomorfo de linfocitos de pequeño y mediano tamaño que presentan núcleo cerebriforme, y c) PL tipo C «tipo células grandes», que se define por un infiltrado denso compuesto fundamentalmente por linfocitos atípicos de gran tamaño3.
ResultadosLos diferentes datos evaluados de los pacientes del estudio se recogen en la tabla 1.
Relación de características clínico-patológicas de los casos incluidos en el estudio
| N.o/sexo/edad | Antecedentes de interés | Lesión elemental | Patrón de distribución e intensidad de afectacióna | Sintomatología asociada | Número máximo de lesiones en un brote | Lesiones residuales | Patrón histológico | Reordenamiento molecular | N.o brotes/año | Duración de la enfermedad | Seguimiento posterior (5 años) |
| 1/mujer/5 años | No | Pápulas parduzcas no confluyentes con ulceración | EESS (++), EEII (+), y tronco (+) | No | 6–10 | Áreas de hiperpigmentación residual y cicatrices varioliformes | PL Tipo Ab | No realizado | 1 único brote | 1 año | Sin complicaciones |
| 2/varón/2 años | No | Pápulas y nódulos rojo-violáceos infiltradas no confluyentes, algunas de ellas con necrosis central | EESS (+++), EEII (++) y tronco (+) | No | 11–20 | Áreas de hipopigmentación residual y cicatrices varioliformes | PL Tipo Ab | No realizado | 3 | Persistencia tras 5 años de seguimiento | Sin complicaciones |
| 3/varón/8 años | No | Pápulas parduzcas no confluyentes con ulceración | EESS (+++), EEII (+++) y tronco (+) | Prurito | 11–20 | Áreas de hiperpigmentación residual | PL Tipo Ab | No realizado | 3 | Persistencia tras 5 años de seguimiento | Sin complicaciones |
| 4/varón/5 años | No | Pápulas parduzcas no confluyentes con ulceración | EESS (++) y EEII (+) | No | 6–10 | Áreas de hiperpigmentación residual y cicatrices varioliformes | PL Tipo Ab | No realizado | 2 | 3 años | Sin complicaciones |
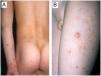
| 5/varón/6 años (fig. 1) | No | Pápulas parduzcas no confluyentes con ulceración | EEII (+++) | No | 11–20 | Áreas de hipopigmentación residual y cicatrices varioliformes | PL Tipo Ab | Monoclonalidad | 2 | Persistencia tras 5 años de seguimiento | Sin complicaciones |
| 6/mujer/10 años | No | Pápulas eritematovioláceas con costra necrótica central | EESS (+++) y EEII (+++) | No | 11–20 | Áreas de hiperpigmentación residual y cicatrices varioliformes | PL tipo Ab | Monoclonalidad | 2 | Persistencia tras 5 años de seguimiento | Desarrollo de lesiones compatibles clínica e histológicamente con pitiriasis liquenoide Sin complicaciones |
| 7/varón/8 años (fig. 2) | PLEVA diagnosticada a los 5 años | Pápulas de consistencia firme eritemato-marronáceas descamativas asociadas a otras lesiones papulosas violáceas ulceradas | EESS (+++), EEII (+++) y tronco (+) | Prurito | 11–20 | Áreas de hiperpigmentación residual y cicatrices varioliformes | PL tipo Ab | No realizado | 3 | Persistencia tras 5 años de seguimiento | Sin complicaciones |
| 8/varón/17 años | No | Pápulas eritematovioláceas induradas e infiltradas, algunas con ulceración central | EESS (+++), EEII (+++), y región facial | No | 11–20 | Áreas de hiperpigmentación residual | PL tipo Ab | Monoclonalidad | 3 | 3 años | Sin complicaciones |
| 9/varón/9 | PLEVA diagnosticada a los 7 años | Pápulas y nódulos indurados, algunas con ulceración central | EESS (++) y EEII (+++) | Prurito | 11–20 | Áreas de hiperpigmentación residual y cicatrices varioliformes | PL tipo Ab | No realizado | 2 | Persistencia tras 5 años de seguimiento | Sin complicaciones |
EEII: extremidades inferiores; EESS: extremidades superiores; PLEVA: pitiriasis liquenoide y varioliforme aguda.
Gradación de mayor o menor concentración de lesiones: +++, más de 10 lesiones; ++, entre 5 y 10 lesiones; +, menos de 5 lesiones.
PL tipo A: papulosis linfomatoide tipo A: infiltrado polimorfo dérmico perivascular superficial y profundo constituido predominantemente por linfocitos grandes atípicos CD 3 y CD30+, CD20, CD15 y EMA – y componente linfoide acompañante CD3+CD20−, y en menor medida, neutrófilos, histiocitos, células plasmáticas y algún eosinófilo.
Un niño de 6 años, sin antecedentes personales ni familiares de interés, acudió a la consulta de dermatología por la presencia desde hacía 2 años de brotes repetidos (2 episodios anuales) de múltiples pápulas rojo-violáceas (de 11 a 20 lesiones) de menos de 1cm de diámetro distribuidas de forma simétrica en miembros inferiores (fig. 1A). Las lesiones papulares eran asintomáticas, infiltradas e induradas al tacto, y algunas de ellas mostraban ulceración central con escara acompañante. Estos brotes mostraban tendencia a la resolución espontánea tras 6–8 semanas de seguimiento, dejando áreas de hipopigmentación residual y discretas cicatrices varioliformes. El paciente no presentaba otras alteraciones clínicas y la exploración física se encontraba dentro de la normalidad.
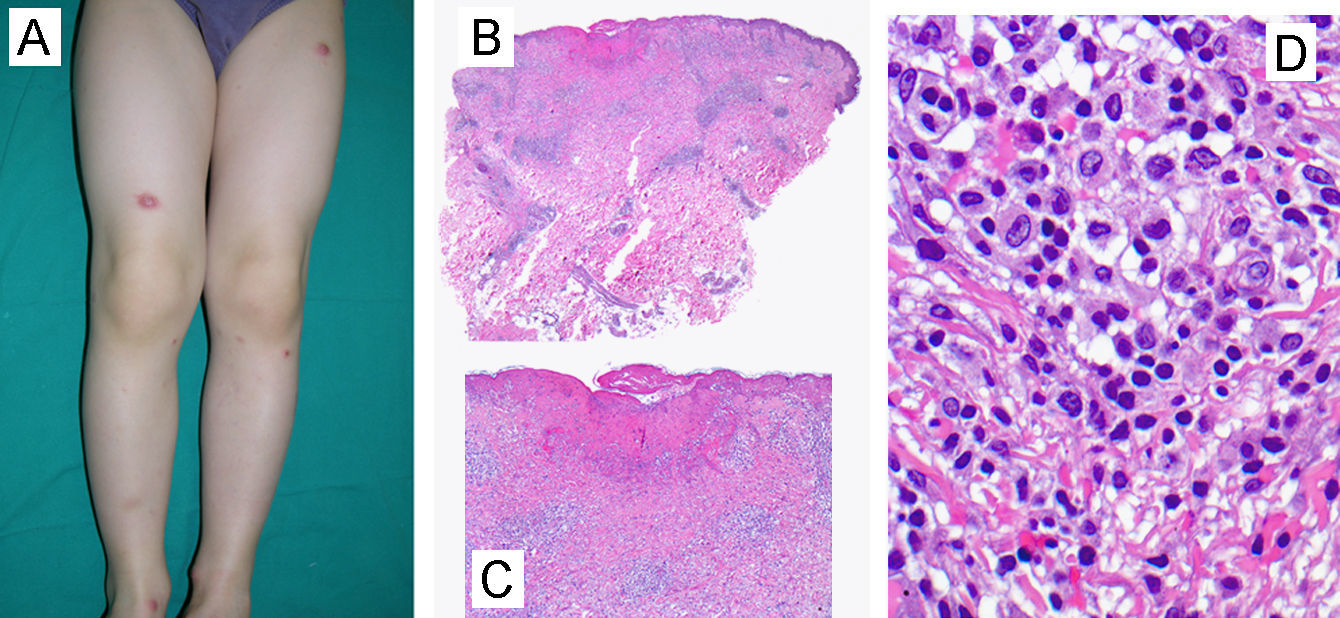
A) Características clínicas: lesiones pápulo-nodulares violáceas distribuidas en extremidades inferiores. (Caso 5, tabla 1) B-D Características histológicas. B) Epidermis ulcerada acompañada de un infiltrado inflamatorio dérmico denso ocupando la dermis media y profunda. (H-E, 40×). C) El infiltrado se compone predominantemente de linfocitos de pequeño tamaño, algunos de ellos atípicos, acompañados de células linfoides de mayor tamaño (H-E, 100×). D) Los linfocitos de mayor tamaño de hábito histiocitoide presentan núcleos vesiculosos (H-E, 400×).
La biopsia de una de estas lesiones papulares mostró un infiltrado dérmico denso de disposición predominantemente perivascular en dermis papilar y reticular, así como a lo largo de la membrana basal, observándose un marcado epidermotropismo y ulceración secundaria de la epidermis (fig. 1B y C). El infiltrado inflamatorio estaba compuesto predominantemente por linfocitos de pequeño tamaño acompañados por otras células linfoides de mayor tamaño con núcleo vesiculoso y nucléolos prominentes que recordaban a las células de Reed-Stemberg (fig. 1D). Asimismo se apreciaban histiocitos, neutrófilos y algún eosinófilo aislado (fig. 1C y D). En el estudio inmunohistoquímico, todas las células linfoides de la lesión fueron positivas para CD3 y CD8, mientras que solo las de mayor tamaño fueron positivas para CD30 en pequeños acúmulos. Este infiltrado fue negativo para EMA, CD15, CD20 y CD79a. El CD68 fue negativo en las células de la lesión pero tiñó los histiocitos acompañantes. El estudio de reordenamiento TCR mediante PCR demostró una población monoclonal de linfocitos T.
Con el diagnóstico de PL tipo A se optó por la abstención terapéutica.
Análisis de los resultadosEl estudio incluyó a 7 niños (77%) y 2 niñas (23%) de edades comprendidas entre 2 y 17 años (mediana, 8 años). Dos de los pacientes desarrollaron las lesiones de PL en el contexto de una pitiriasis liquenoide aguda (PLEVA) confirmada histológicamente e iniciada en años previos.
Clínicamente, los 9 casos (100%) se presentaron en forma de pápulas rojo-marronáceas y/o eritematovioláceas leve o moderadamente infiltradas. El número máximo de lesiones por brote fue variable, siendo entre 11 y 20 en 7 casos (77%), y menor de 10 en los 2 casos restantes. Las lesiones presentaron prurito asociado en un 33% de los casos y se distribuyeron en orden decreciente de frecuencia en extremidades inferiores (9 casos, 100%), extremidades superiores (8 casos, 88%), tronco (4 casos, 44%) y en región facial (1 caso, 10%). De forma llamativa las lesiones se concentraron en mayor número en las extremidades en todos los casos, mientras que en aquellos casos en los que se produjo alteración de la cara y del tronco, esta ocurrió en forma de lesiones aisladas (tabla 1). La resolución de las lesiones fue espontánea en todos los casos, con hiperpigmentación (7 casos, 77%) o hipopigmentación postinflamatoria (2 casos, 23%). El desarrollo de cicatrices varioliformes residuales se objetivó en 7 pacientes (77% de los casos).
El número de brotes/año fue variable, oscilando entre 2 y 3 al año en 8 pacientes (90%). Excepcionalmente 1 paciente mostró un único brote de lesiones que posteriormente no se acompañó de nueva clínica cutánea en el seguimiento periódico. En 6 de los casos (66%), la actividad de la enfermedad no cesó tras los 5 años de seguimiento. Dos pacientes (22%) mostraron remisión de la enfermedad a los 3 años de controles periódicos. Uno de los casos (12%) solo presentó actividad el primer año de seguimiento.
Ninguno de los 9 casos presentó complicaciones relevantes en los 5 años de vigilancia posteriores. No obstante, 1 de los pacientes desarrolló en el seguimiento lesiones compatibles clínica e histológicamente con pitiriasis liquenoide crónica.
Desde el punto de vista histológico, los 9 casos (100%) presentaron un patrón compatible con PL tipo A. El estudio molecular del reordenamiento de RCT mostró monoclonalidad en los 3 casos en los que fue realizado.
DiscusiónLa PL es una enfermedad de etiología desconocida que actualmente se incluye dentro de los linfomas cutáneos de células T CD30+ de curso indolente, según la clasificación de la European Organization for Research and Treatment of Cancer (EORTC)2.
Se trata de un cuadro que generalmente aparece en adultos de edades comprendidas entre los 40 y los 60 años, sin mostrar predilección por sexo o raza3. Clínicamente se manifiesta con el desarrollo de múltiples brotes de lesiones pápulo-nodulares y con menor frecuencia vesículo-pustulosas, localizadas principalmente en tronco y extremidades, que muestran una tendencia a la resolución espontánea, dejando en algunos casos una cicatriz residual3. La duración de estos brotes varía entre semanas y meses, y el intervalo libre de enfermedad entre ellos oscila de semanas a años. Característicamente en un gran porcentaje de pacientes este intervalo libre de lesiones se va alargando progresivamente hasta la ausencia de nuevos brotes. Se trata de un proceso de pronóstico excelente, con una supervivencia global a los 5 años del 100%3.
Desde el punto de vista histológico la PL muestra 3 patrones diferentes que no muestran correlación con el pronóstico a largo plazo. La forma más frecuente, con un 75% de los casos, es el patrón tipo A o histiocitoide, que se caracteriza por un infiltrado mixto compuesto mayoritariamente por linfocitos de pequeño tamaño junto a múltiples linfocitos atípicos de gran tamaño o células histiocitoides. Hasta un 4% de los casos de PL muestran el patrón tipo B o «tipo micosis fungoide», que se caracteriza por un infiltrado monomorfo de linfocitos de pequeño y mediano tamaño que presentan núcleo cerebriforme. Por último, la PL tipo C «tipo células grandes», que se caracteriza por un infiltrado denso compuesto fundamentalmente por linfocitos atípicos de gran tamaño, aparece hasta en un 13% de los casos3.
La principal complicación de este cuadro es el hecho de que entre el 5 y el 20% de los pacientes con PL desarrollan en su seguimiento a largo plazo un nuevo proceso linfoproliferativo3, principalmente una micosis fungoide, y con menos frecuencia una enfermedad de Hodking o un linfoma anaplásico de célula grande CD30+ cutáneo y sistémico4.
El debut de este proceso en la infancia, considerando en este grupo a todos aquellos pacientes menores de 18 años, es excepcional. Hasta la actualidad se han descrito aproximadamente unos 70 casos de PL en edad infantil5–10. La mayoría de los casos proceden de tres grandes series de 35, 14 y 10 pacientes publicadas por Van Neer5, Nijsten6 y De Souza7, respectivamente.
La PL infantil, que supone del 2 al 9% del global del as PL3, aparece con una frecuencia discretamente superior en varones, de edades comprendidas entre 1 y 17 años (mediana de 8 años)5–10. Estos datos epidemiológicos coinciden con los reflejados en nuestra serie, en la que el 77% de los pacientes fueron varones con una edad media de 7,6 años. Los varones muestran una tendencia a presentar un debut más precoz del cuadro clínico (mediana: 5,5 años) comparado con el sexo femenino (mediana de 12 años)5–10. Como antecedentes previos de interés, resulta llamativo el hecho de que 2 pacientes de nuestra serie debutaron en los años previos con lesiones clínica e histológicamente compatibles con pitiriasis liquenoide, para posteriormente desarrollar el cuadro linfoproliferativo T CD30+ (fig. 2A y B).
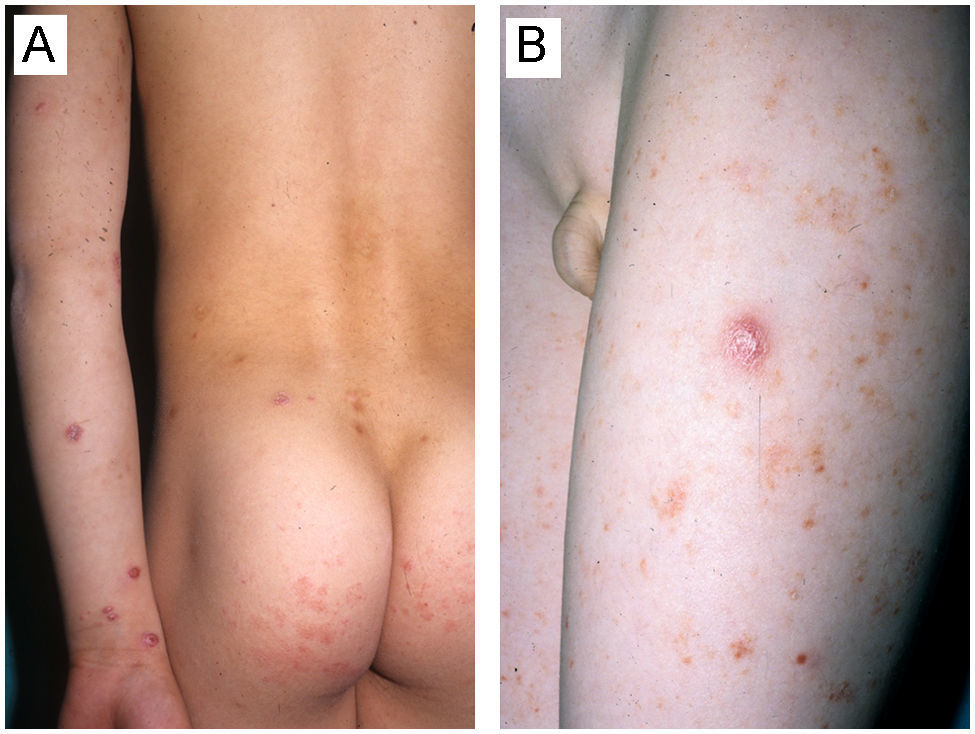
A y B) Varón de 8 años que, en el contexto de un cuadro conformado por lesiones eritemato-descamativas no infiltradas predominantemente afectando el tronco clínica e histológicamente compatibles con pitiriasis liquenoide, desarrolló posteriormente lesiones pápulo-nodulares infiltradas violáceas compatibles con papulosis linfomatoide afectando a extremidades superiores, inferiores y en menor medida tronco (Caso 7, tabla 1).
Las características clínicas de la PL infantil no parecen diferir sustancialmente de la forma adulta de la PL. Esta se caracteriza por la existencia de brotes recurrentes de pápulas y/o nódulos afectando de forma simétrica y en mayor intensidad a las extremidades (88% de los casos en EE superiores e inferiores), y en menor frecuencia e intensidad al tronco (en 44% de los casos) a la región facial (10% de los casos)5–10. La variante localizada y la folicular de PL, que son formas de presentación excepcionales de esta enfermedad, se describen con mayor frecuencia en la forma infantil de PL3,8. Por el contrario, la afectación de la mucosa oral, descrita en la edad adulta, no ha sido reportada en ningún caso en edades infantiles11. Las lesiones son frecuentemente asintomáticas; sin embargo, en nuestra serie un 33% presentaron prurito asociado, fenómeno que se describe hasta un 40% de casos publicados en la literatura5–10. Típicamente, estos episodios recurrentes de lesiones de PL presentan una duración que oscila entre 2 y 8 semanas, seguido por la resolución espontánea de las lesiones. El desarrollo de cicatrices residuales es relativamente frecuente, apareciendo en nuestro estudio hasta en el 77% de los casos, dato concordante con los datos previamente publicados5–10. La duración de la enfermedad fue variable, pero con mayor frecuencia permaneció activa tras los 5 años de seguimiento del estudio.
Histológicamente, la PL infantil puede presentar cualquiera de los tres patrones descritos anteriormente. Sin embargo, en las series publicadas previamente el patrón que aparece con mayor frecuencia es el tipo A o histiocitoide5–10. En nuestro estudio el 100% de los casos presentan este patrón de PL tipo A, con lo que se concluye que este tipo morfológico es la variante con la que, salvo excepciones, la PL se manifiesta histopatológicamente en edades juveniles. Inmunohistoquímicamente las células atípicas son CD2, CD3 y CD30 positivas. Característicamente la positividad para CD30 aparece en células atípicas grandes aisladas y formando pequeños grupos. No obstante, a diferencia de la forma adulta en la que los linfocitos que predominan en el infiltrado son CD4+ CD8−3, en la variante infantil aparece con mayor frecuencia un infiltrado compuesto mayoritariamente por linfocitos T citotóxicos CD8+5,6,12. Los resultados del estudio de clonalidad celular son variables y carecen de relevancia pronóstica3,5–10. Sin embargo, el carácter monoclonal de nuestros 3 pacientes analizados fue de utilidad para corroborar el diagnóstico de PL.
El análisis conjunto de las 6 grandes series de PL infantil5–10, muestra la existencia de solo 3 casos de pacientes que desarrollan un nuevo proceso linfoproliferativo en su seguimiento a largo plazo. Los 3 pacientes, que pertenecen a la serie de Nijsten6, desarrollaron un linfoma no Hodking tras un intervalo de tiempo desde el debut de la PL que osciló entre 1 mes y 17 años. En nuestro estudio ningún caso presentó complicaciones secundarias en los posteriores 5 años de seguimiento.
La PL de la infancia debe ser diferenciada principalmente de las picaduras de artrópodos y de la pitiriasis liquenoide. Las picaduras por insectos muestran un curso más corto y autolimitado de las lesiones, y su histología revela un infiltrado inflamatorio mixto en disposición en pirámide invertida con su base dispuesta paralela a la epidermis en la que predominan los eosinófilos. Clásicamente, la pitiriasis liquenoide se diferencia de la PL desde el punto de vista clínico e histopatológico. Esta entidad se manifiesta en forma de brotes de múltiples pápulas y placas eritemato-descamativas no induradas ni infiltradas de aparición brusca. Desde el punto de vista histológico la pitiriasis liquenoide se caracteriza por una epidermis paraqueratósica con degeneración vacuolar de la membrana basal asociada a la presencia de múltiples queratinocitos necróticos y a un infiltrado inflamatorio liquenoide compuesto principalmente por linfocitos T maduros, que inmunohistoquímicamente son CD30 negativos o muestran una positividad leve y dispersa. Sin embargo, hay que tener presente que en ocasiones es muy difícil establecer una barrera clara entre la PLEVA y la PL, ya que existen casos en los que ambas entidades se solapan clínica e histológicamente. En estos casos, el patrón clínico y evolutivo del cuadro permitirá establecer el diagnóstico definitivo (tabla 2). Por otro lado, estas dificultades en el momento de diferenciar ambas entidades, unido a la elevada frecuencia de aparición concomitante de pitiriasis liquenoide y PL descrito en nuestra serie, nos plantean la posibilidad de que ambas entidades podrían estar formando parte un espectro clínico común.
Características diferenciales entre papulosis linfomatoide y pitiriasis liquenoide
| Papulosis linfomatoide | Pitiriasis liquenoide | |
| Inicio del cuadro | Insidioso | Brusco |
| Número de lesiones en cada brote | Discreto (entre 1 y 20) | Múltiple (más de 20) |
| Características de la lesión | Pápulas y nódulos que asocian en ocasiones una escara necrótica central | Placas eritematodescamativasa |
| Lesiones induradas al tacto e infiltradas a planos profundos | Lesiones no induradas ni infiltradas a planos profundos | |
| Patrón de distribución de las lesiones | Predominantemente en EESS y EEII | Predominantemente en tronco |
| Evolución del cuadro | ||
| Duración de las lesiones | Resolución en 3 a 4 semanas | Resolución en 1 a 2 semanas |
| individuales | ||
| Alteración residual tras resolución de lesiones | Cicatrices varioliformes residuales | Áreas de hipo/hiperpigmentación residual |
| Duración de los brotes | Años | PLEVA, semanas a meses |
| PLC, meses a años | ||
| Hallazgos histológicos | Tipo A: presencia de células linfocíticas atípicas con núcleo bizarro acompañadas de un infiltrado neutrofílico, con mínima afectación de la epidermis | PLEVA: infiltrado linfocítico severo, con rara presencia de algún linfocito atípico de forma aislada. Epidermis afecta, con degeneración hidrópica de la membrana basal, queratinocitos necróticos |
| Tipo B: células mononucleares hipercromáticas cerebriformes dispuestas en banda en la membrana basal, con cierta degeneración hidrópica | PLC: infiltrado linfocítico leve, ausencia de linfocitos atípicos, mínima degeneración vacuolar de la membrana basal | |
| Tipo C: PL «tipo células grandes», se define por un infiltrado denso compuesto fundamentalmente por linfocitos atípicos de gran tamaño | ||
| Positividad para el marcador CD 30 | Presente (ausente en un 5% de casos) | Ausenteb (presente en un 5–10% de casos) |
| Estudio de reordenamiento molecular | Monoclonalc (policlonal en un 30–40% de casos) | Policlonalc (monoclonal en un 10% de casos) |
| Potencial de malignidad | Ocasional | Excepcional |
EEII: extremidades inferiores; EESS: extremidades superiores; PLC: pitiriasis liquenoide crónica; PLEVA: pitiriasis liquenoide y varioliforme aguda; PL: papulosis linfomatoide.
La infrecuente variante ulcero-necrótica febril de Mucha Habermann se caracteriza por placas ulceradas localizadas eminentemente en tronco asociado a un proceso febril.
En algunos casos, el marcador CD30 resulta positivo y el estudio molecular muestra carácter monoclonal. En estos casos, el patrón clínico-evolutivo será fundamental establecer el diagnóstico definitivo.
La existencia de reordenamiento monoclonal de los linfocitos T es un dato de ayuda para apoyar el carácter neoplásico de un proceso y carece de relevancia pronóstica. No obstante, las características clínicas y evolutivas del cuadro problema resultan de mayor relevancia en el establecimiento del diagnóstico definitivo.
El tratamiento de la PL infantil en muchos casos no es necesario, ya que la duración del período de actividad de la enfermedad no se ha asociado a un peor pronóstico. A su vez ninguno de los tratamientos actualmente disponibles resulta en la remisión completa de la enfermedad, consiguiendo únicamente una resolución parcial y temporal del cuadro. Por ello, la intervención terapéutica solo está indicada por la existencia de sintomatología asociada, tal como prurito, o por motivos cosméticos en aquellos casos de brotes de muchas lesiones. En el caso de la PL infantil, el principal problema radica en la ansiedad que genera a los padres la presencia de estas lesiones en sus hijos. Las terapias descritas en la PL infantil incluyen la administración tópica o sistémica de corticosteroides, antibióticos orales (macrólidos principalmente)13,14, PUVA terapia15 y UVB13.
Comunicamos esta serie de PL infantil porque se trata de una enfermedad rara y por tanto, mal caracterizada en este grupo de edad. Nuestros hallazgos clínico-patológicos mostraron concordancia con los publicados previamente. Es importante tener presente el hecho de que pacientes con PL pueden asociar un proceso compatible con pitiriasis liquenoide previa o posteriormente, tal y como ocurre en nuestra serie. La elevada frecuencia de asociación entre ambas entidades que se describe en nuestra serie sugiere que estas enfermedades representen polos opuestos de un espectro clínico y patológico común. Desde el punto de vista histológico los resultados del estudio permiten establecer a la variante tipo A de PL como el patrón histopatológico característico de la PL en la infancia. Por otro lado, obsevamos que las lesiones de PL en la infancia se resuelven dejando una alteración transitoria de color de la piel en todos los casos, principalmente en forma de hiperpigmentación, acompañada de lesiones cicatriciales permanentes en más de la mitad de los pacientes. Por último queremos destacar, según se desprende de las series y casos publicados y de nuestra propia experiencia, que el desarrollo de otros procesos linfoproliferativos es menos frecuente en la PL infantil que en la variante adulta y que se manifiesta, según la literatura, principalmente en forma de linfoma no Hodking.


