


Maculopapular cutaneous mastocytosis, previously known as urticaria pigmentosa, is the most common cutaneous expression of indolent systemic mastocytosis (SM) in adults. This condition usually does not require specific treatment.1 However, SM can be associated with other hematologic malignancies.2 We present the case of a patient with indolent SM who developed essential thrombocythemia during clinical follow-up.
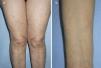
A 70-year-old woman with no relevant past history except dyslipidemia was referred to our dermatology department in 2005 for assessment of brownish papules measuring 3-5mm in diameter on the limbs and upper chest that had appeared gradually over the previous 5 years (Fig. 1). On examination, the Darier sign was observed. The patient reported neither pruritus nor symptoms associated with mast cell degranulation. Skin biopsy revealed mast cell proliferation in the upper dermis. Serial bone radiography and abdominal ultrasound were normal. Complete blood count and serum biochemistry were normal except for tryptase levels (124μg/L; normal<11.4μg/L). The patient was referred to the hematology department and underwent bone marrow aspiration, which revealed the presence of atypical mast cells consistent with a diagnosis of SM (flow cytometry detected 0.2% mast cells, 100% with pathologic phenotype CD2+/CD25+). Polymerase chain reaction detected the presence of the c-kit D816V mutation. With a diagnosis of indolent SM, the patient was once again referred to the dermatology department without treatment. The platelet count increased gradually from 2010 to 2013 until it reached 741×109/L, at which point the patient was once again referred to the hematology department for assessment of thrombocytosis. The patient was diagnosed with mastocytosis associated with essential thrombocythemia–type myeloproliferative neoplasm. Treatment was initiated with oral hydroxyurea (500mg, 3 d/wk) and oral clopidogrel (75mg, every 48h). The patient has not developed any symptoms associated with mast cell degranulation. The lesions on the arms and upper chest have gradually resolved and only a few lesions persist on the thighs. Platelet count has remained below 700×109/L and serum tryptase levels remain at around 100μg/L.
SM is classified as indolent SM, smoldering SM, aggressive SM, SM with associated clonal hematologic non-mast-cell lineage disease, mast cell leukemia, and mast cell sarcoma.3 Indolent SM is the most common form, but it does not usually cause symptoms derived from mast cell infiltration and does not require cytoreductive therapy.1 The second most common form is SM associated with hematologic disease, which accounts for between 21% and 44% of SM cases.2 The hematologic disorders most frequently associated with SM are myeloproliferative syndromes (45% of cases), chronic myelomonocytic leukemia (29%), myelodysplastic syndromes (23%), and acute leukemia (3%).2 Essential thrombocythemia is classified as a myeloproliferative syndrome. The association of SM with essential thrombocythemia is very rare. In a series of 123 patients with SM associated with hematologic disease, essential thrombocythemia accounted for only 6 cases.2 Besides these 6 patients, the reviewed literature only contains a few isolated case reports describing the association between maculopapular mastocytosis and essential thrombocythemia.4–10
Although the pathophysiologic relationship between mast cell proliferation and associated hematologic malignancies is not well established, some lineage distribution studies of the c-kit D816V mutation suggest the existence of a common pluripotent hematopoietic stem cell in most patients.2 In some cases, however, it is possible that the 2 clonal hematologic disorders may develop coincidentally in the same patient.2
When we talk about SM, we tend to think of asymptomatic mast cell infiltration in bone marrow or the rare aggressive forms of mastocytosis. However, we must also be aware of the possibility of association with other hematologic malignancies, as the case of essential thrombocythemia in our patient illustrates. The prognosis of indolent mastocytosis associated with clonal hematologic non-mast-cell lineage disease depends on the type of associated hematologic disorder. Cutaneous manifestations of mastocytosis can therefore be considered possible markers of a more severe associated hematologic disorder that may require specific treatment.
All adult patients with mastocytosis should undergo a bone marrow study that includes mutational analysis and mast cell immunophenotyping. These patients require clinical follow-up not only because of the risk of developing aggressive forms of SM but also because of the risk of associated hematologic malignancies.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Marcoval J. Mastocitosis asociada a trombocitemia esencial. Actas Dermosifiliogr. 2019;110:63–64.


