



















Las malformaciones vasculares (MV) son errores innatos en el desarrollo embriológico de los vasos sanguíneos que están presentes siempre desde el nacimiento. Por ello deben diagnosticarse en la infancia y, en los casos en que sea necesario, tratarlas para evitar complicaciones posteriores. La tendencia actual es a clasificar estas lesiones en cuanto al flujo que presentan y en cuanto al tipo de vaso predominante en las mismas.
Dada la complejidad que presentan estas lesiones, y en muchos casos su poca frecuencia, deben abordarse desde un punto de vista multidisciplinario en centros de anomalías vasculares.
Además, la asociación de malformaciones vasculares con cuadros sindrómicos está cada vez mejor definida, lo que hace necesario su conocimiento para un mejor abordaje de estos enfermos.
Vascular malformations are inborn errors of vascular embryogenesis present at birth that should be diagnosed in childhood and, when necessary, treated to prevent later complications. The current trend is to classify these lesions according to flow characteristics and the predominant type of vascular channel affected.
Given the complexity, and in many cases, the rarity, of vascular malformations, they should be managed by multidisciplinary teams at vascular anomalies centers.
Furthermore, because the association between vascular malformations and certain syndromes is becoming increasingly recognized, a better understanding of these lesions will help to improve overall patient management in this setting.
Si existe un grupo patológico dentro de la medicina que ha causado confusión a lo largo de la historia es el de las anomalías vasculares. De hecho el principal problema que ha existido siempre para el avance en el conocimiento de estas lesiones ha sido unificar su nomenclatura.
El paso más importante en este sentido fue el trabajo de Mulliken y Glowacki en 19821, que propusieron una nueva clasificación de las anomalías vasculares basada en criterios clínicos, radiológicos, hemodinámicos e histopatológicos. Este trabajo es el punto de partida para la clasificación de las anomalías vasculares más aceptada actualmente, que es la de la Internacional Society for Study of Vascular Anomalies (ISSVA). De este modo en las últimas dos décadas los avances en las bases genéticas, los mecanismos fisiopatológicos, el diagnóstico y el tratamiento de las anomalías vasculares han sido espectaculares2.
Definición, clasificación e importanciaLa clasificación de la ISSVA divide las lesiones vasculares benignas fundamentalmente en dos grupos: tumores —dentro de los cuales el más frecuente es el hemangioma infantil— y las malformaciones vasculares.
Las malformaciones vasculares (MV) son errores innatos en el desarrollo embriológico de los vasos sanguíneos que están presentes siempre desde el nacimiento, aunque a veces se manifiesten o den problemas más tarde. De forma general presentan un crecimiento gradual, proporcionalmente mayor al crecimiento del paciente, que es más significativo en la pubertad.
La clasificación de las malformaciones vasculares de la ISSVA (Budapest, 1992), las divide según el vaso predominante (tabla 1). Pero hay otra forma de clasificarlas muy importante desde el punto de vista funcional, y con implicaciones tanto diagnósticas como terapéuticas, basada en el flujo de las lesiones3 (tabla 1).
Clasificación de las malformaciones vasculares según su flujo
| Clasificación según su flujo |
| Flujo lento |
| Malformaciones capilares (MC) |
| Malformaciones venosas (MV) |
| Malformaciones linfáticas (ML) |
| Flujo rápido |
| Malformaciones arteriales (MA) |
| Malformaciones arteriovenosas (MAV) |
| Clasificación de laInternational Society for Study of Vascular Anomalies(ISSVA) |
| Tumores vasculares |
| Hemangiomas infantiles |
| Hemangiomas congénitos (rápidamente RICH o no involutivos NICH) |
| Angioma en penacho (con o sin Kasabach-Merrit) |
| Hemangioendotelioma kaposiforme (con o sin Kassabach-Merritt) |
| Hemangioendotelioma de células fusiformes |
| Otros hemangioendoteliomas |
| Epitelioide, retiforme, polimorfo, tumor Dabska, linfangioendoteliomatosis, etc. |
| Tumores vasculares dermatológicos adquiridos |
| Granuloma piogénico, hemangioma hemosiderótico, hemangioma glomeruloide, hemangioma microvenular, etc. |
| Malformaciones vasculares |
| Malformaciones de bajo flujo |
| Malformaciones capilares (CM) |
| Mancha en vino de Oporto, telangiectasia, angioqueratoma |
| Malformaciones venosas (VM) |
| Esporádicas comunes, síndrome de Bean, MV cutáneas y mucosas familiares, malformación glomovenosa, síndrome de Maffucci |
| Malformaciones linfáticas (LM) |
| Linfedema primario. Malformaciones linfáticas microquísticas y macroquísticas |
| Malformaciones vasculares combinadas complejas |
| CVM, CLM, LVM, CLVM |
| Malformaciones de flujo alto |
| Malformaciones arteriales, fístula arteriovenosa o malformación arteriovenosa |
| Malformaciones vasculares combinadas complejas |
| AVM-LM, CM-AVM |
En los primeros estudios realizados sobre las malformaciones vasculares se decía que en ellas no hay proliferación celular, sino que eran vasos displásicos con recambio endotelial normal. Sin embargo, este hecho hoy está en entredicho, sobre todo en las malformaciones arteriovenosas (MAV) que, en ocasiones, presentan un crecimiento de rango tumoral e inducen proliferación de células endoteliales4.
Es muy importante conocer estas lesiones, ya que pueden presentarse de forma aislada o asociarse a síndromes más complejos con afectación de otros órganos. Los síndromes asociados a malformaciones vasculares en la infancia se abordarán en la segunda parte de este trabajo.
Malformaciones capilaresProbablemente las malformaciones capilares (MC) constituyen el grupo más heterogéneo dentro de la clasificación de la ISSVA de las malformaciones vasculares, lo que ha sido criticado en algunos trabajos5.
Llamamos MC a aquellas en que los vasos predominantes son las arteriolas o vénulas poscapilares, es decir vasos de pequeño grosor y flujo lento.
Si exceptuamos la mancha salmón se producen en el 0,3% de la población y afectan a ambos sexos por igual; aunque generalmente son esporádicas se han descrito casos familiares.
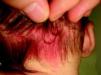
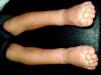
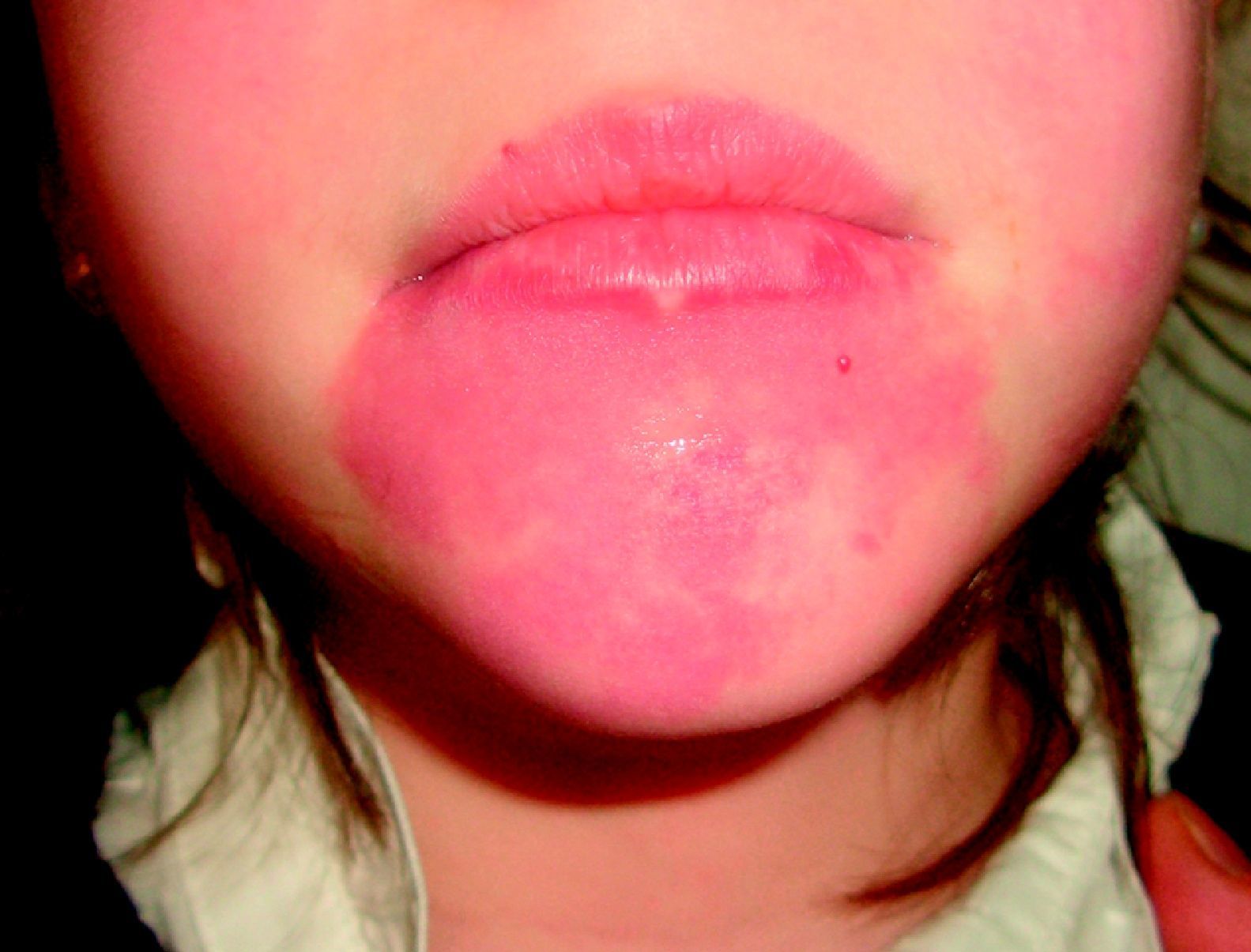
Mancha salmónLa MC que vemos con mayor frecuencia es la mancha salmón, que puede afectar hasta la mitad de los neonatos6,7. Es una mácula de tonalidad rosada que suele afectar la zona occipital (picotazo de cigüeña [stork-bite]) o el área glabelar (beso de ángel), pero también pueden afectar los párpados, la nariz, el labio superior o la región sacra (fig. 1). Suelen ser lesiones aisladas, y aunque la mayoría de ellas desaparecen, algunas persisten durante toda la vida. Es importante no confundirlas con lesiones precursoras de hemangiomas, por lo que en los primeros meses de vida es aconsejable realizar un seguimiento. También en ocasiones en que tienen una tonalidad más marcada de lo normal pueden confundirse con manchas en vino de Oporto; la evolución nos dará el diagnóstico diferencial.
No suelen tratarse, pero responden muy bien al tratamiento con láseres específicos vasculares, como el láser de colorante.
Estas lesiones que raramente se biopsian, dada su benignidad y resolución espontánea en la mayoría de los casos, son de gran interés porque histopatológicamente son similares a las manchas en vino de Oporto de las que hablaremos más adelante, pero a diferencia de estas, que no desaparecen nunca, la mayoría se resuelven en unos meses. Conocer los mecanismos fisiopatológicos que rigen la involución de estas lesiones podría ayudarnos a tratar las MC que no desaparecen sin tratamiento.
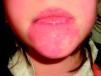
Mancha en vino de OportoEstas lesiones están presentes desde el nacimiento, y después de los primeros meses de vida se estabilizan delimitando el territorio que van a ocupar. Algunas de estas lesiones presentan de forma temporal cierto grado de aclaramiento por mecanismos no bien conocidos.
Suelen manifestarse como máculas rojizas de mayor o menor intensidad, y pueden afectar cualquier parte de la superficie corporal, aunque la localización más frecuente es la facial (fig. 2). Pueden formar parte de síndromes más complejos (tabla 2).
Cuadros síndromicos asociados a malformaciones capilares tipo mancha en vino de Oporto
| Síndrome Sturge-Weber |
| Síndrome de Klippel-Trenaunay |
| Síndrome de Parkes Weber |
| Facomatosis pigmentovasculares |
| Síndrome Proteus |
| Síndrome de Cobb |
| Síndrome de Bannayan-Riley-Ruvalcaba |
| Síndrome de Servelle-Martorell |
| Síndrome de Beckwith-Wiedemann |
| Síndrome Robert (pseudotalidomida) |
| Síndrome trombopenia-ausencia radio (TAR) |
| Síndrome de von Hippel-Lindau |
| Síndrome Rubinstein-Taybi |
| Enfermedad de Coats |
| Síndrome CLAPO |
| Síndrome CLOVES |
| RASA-1: CM-AVM |
| Angiomiolipomas y MVO |
| Macrocefalia-CM o CMTC |
El estudio histopatológico de estas lesiones nos muestra un aumento del número de vasos localizados en la dermis papilar y reticular, junto a un aumento de la ectasia vascular, debido a una falta de fibras nerviosas de estos vasos.
La evolución natural de estas lesiones es a una progresiva profundización, engrosamiento y oscurecimiento debido, en gran parte, a una dilatación secuencial de los vasos que la componen. Este hecho, mucho más patente en la cara, hace que estas lesiones a partir de la tercera o cuarta década se acompañen de hipertrofia cutánea y formación de nódulos. Esta nodularidad restringida a la cara, que no se ve en la infancia, ha hecho a algunos autores postular que se trata realmente de lesiones hamartomatosas8.
El diagnóstico es fundamentalmente clínico9, pero hay que tener en cuenta que sobre todo en la infancia la diferencia con MAV en fase estable es imposible, incluso con la ayuda de pruebas de imagen o de flujo.
La localización de estas lesiones en algunas áreas puede ser un marcador de malformaciones asociadas. Esto es sobre todo importante cuando estas lesiones asientan en la primera rama del trigémino (síndrome de Sturge Weber)10, en la zona lumbar (disrafismo espinal)11,12, en la zona dorsolumbar (síndrome de Cobb), o en el labio inferior (síndrome de CLAPO).
El tratamiento de elección son los láseres vasculares, sobre todo el láser de colorante pulsado13,14, aunque se han descrito resultados satisfactorios con láser de Nd:YAG de pulso largo, KTP láser y con fuentes de luz pulsada intensa15,16. Pero a pesar de los avances que se han producido en estas fuentes de luz, no conseguimos eliminar de forma completa más que un 25-30% de estas lesiones. Para intentar mejorar este tratamiento se han modificado los sistemas de los láseres vasculares añadiendo frío al tiempo del disparo de láser, y sobre todo con láseres de 755nm17 o creando sistemas duales con aplicación de láser de colorante y Nd:YAG de pulso largo18, que parece que han mejorado estos porcentajes. Otras alternativas que se han intentado es asociar el tratamiento con imiquimod19 o con antiangiogénicos tópicos como la rapamicina20,21.
Un dilema que se ha planteado desde hace años es el momento adecuado para comenzar el tratamiento de estas lesiones. En la actualidad se plantea que dado que con el tiempo se hacen más profundas e inducen hipertrofia en la piel, se beneficiarían de tratamientos tempranos. Estos tratamientos en muchos centros se realizan bajo sedación con anestesia general, lo cual plantea varios problemas: por una parte la necesidad de realizarlos en quirófano, con el considerable aumento de coste sanitario y, por otra parte, el riesgo que puede suponer la realización de sedaciones sucesivas para tratar un problema esencialmente cosmético, a pesar de las implicaciones psicológicas que puede acarrear para el paciente y la familia22–24. Los avances en anestesia pediátrica hacen que alternativas como el óxido nitroso o la sedación fuera de quirófano permitan el tratamiento de superficies mayores.
Las manchas en vino de Oporto (MVO), sobre todo cuando afectan a las extremidades, pueden acompañarse de hipertrofia de los tejidos blandos subyacentes. Es muy importante conocer esta variante de hemihipertrofia denominada «hemihipertrofia estable asociada a malformaciones capilares», porque su curso es totalmente benigno, de modo que persiste durante toda la vida esa diferencia relativa que aparece en el nacimiento. Su diagnóstico diferencial con otras entidades que cursan con sobrecrecimiento, como el síndrome de Klippel-Trenaunay, en ocasiones es muy difícil, y los errores diagnósticos son frecuentes.
Hay un grupo de MVO que son adquiridas, aunque con unas características idénticas a las lesiones congénitas25.
En los últimos años se ha descrito la asociación de lesiones tipo MVO de pequeño tamaño, múltiples, relacionadas con MAV cerebrales, y todo ello vinculado con el gen RASA-126.
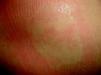
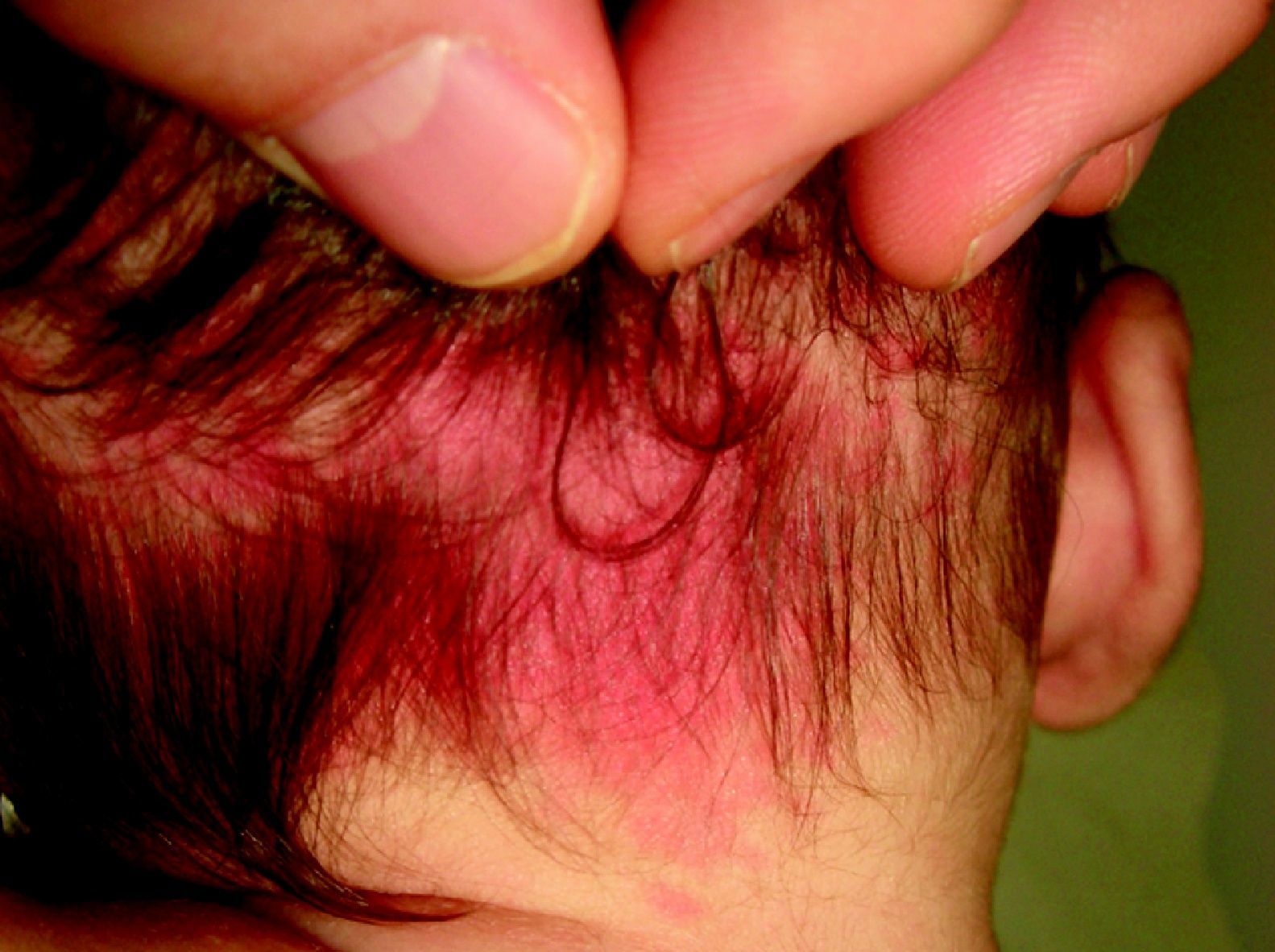
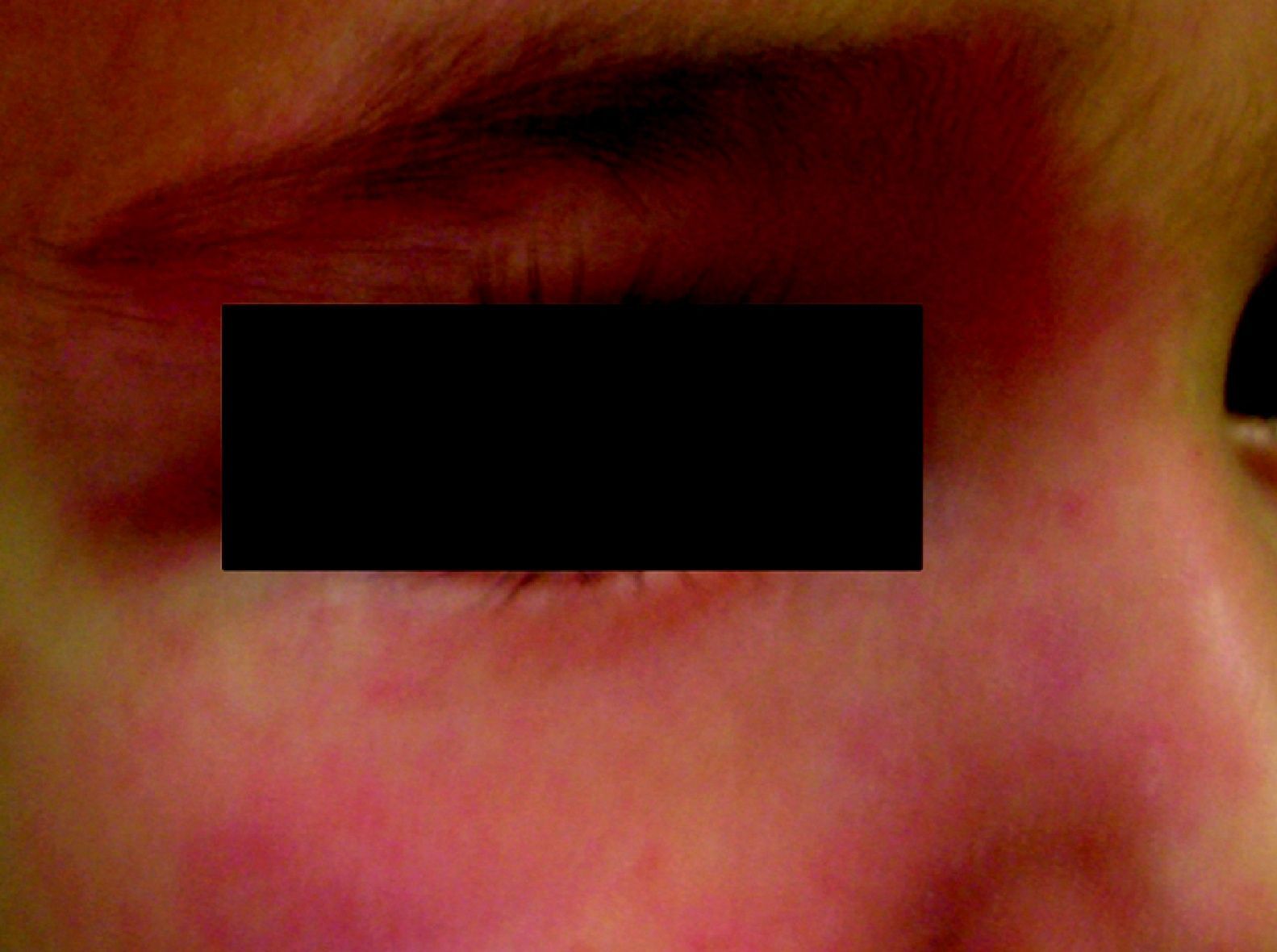
Nevus anemicusTambién llamado nevus nervioso, secundario o funcional. Es una anomalía localizada, congénita, no familiar caracterizada por máculas hipopigmentadas de contornos irregulares que se agrupan formando placas y se localizan sobre todo en la cara anterior del tórax. Esta lesión tiende a aparecer al nacimiento o en los primeros días de vida, y es más frecuente en mujeres. Para confirmar el diagnóstico podemos frotar un poco la zona periférica y veremos que el eritema que se induce en ella no aparece en la zona hipopigmentada, haciendo esta más marcada27 (fig. 3).
Descrito por Van Hömer en 1906, refería una disminución del número y grosor de los vasos sanguíneos en el área afecta, por lo que postuló que podría producirse por aplasia parcial de los vasos dérmicos. Posteriormente se demostró que el número de vasos en esta zona era normal, pero con una sensibilidad aumentada a las catecolaminas, es decir, una estimulación vasoconstrictora permanente28. Cuando se biopsia un nevus anémico y se injerta en otra zona de piel permanece la palidez de la misma. También se ha demostrado que en esta zona hay una disminución de la sensibilidad retardada, porque estos vasos no responden de forma adecuada a las citocinas proinflamatorias29.
Normalmente es un hecho aislado en individuos sanos, pero se ha descrito asociado a pacientes con neurofibromatosis y a pacientes con nevus flammeus30, algunos de ellos con síndrome de Sturge-Weber asociado. Cuando a estos dos se asocia la mancha mongólica conforman la facomatosis pigmento vascular de tipo iib. También se ha asociado con nevus spilus y linfedema primario, con alopecia universal, onicodistrofia, vitíligo extenso y atopia.
El diagnóstico diferencial debe realizarse con la pitiriasis versicolor, el vitíligo, el nevus depigmentosus, las máculas hipopigmentadas de la esclerosis tuberosa y de la lepra.
Esta entidad no está incluida en la clasificación de la ISSVA dentro de las MC, pero probablemente debería formar parte de este grupo.
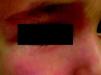
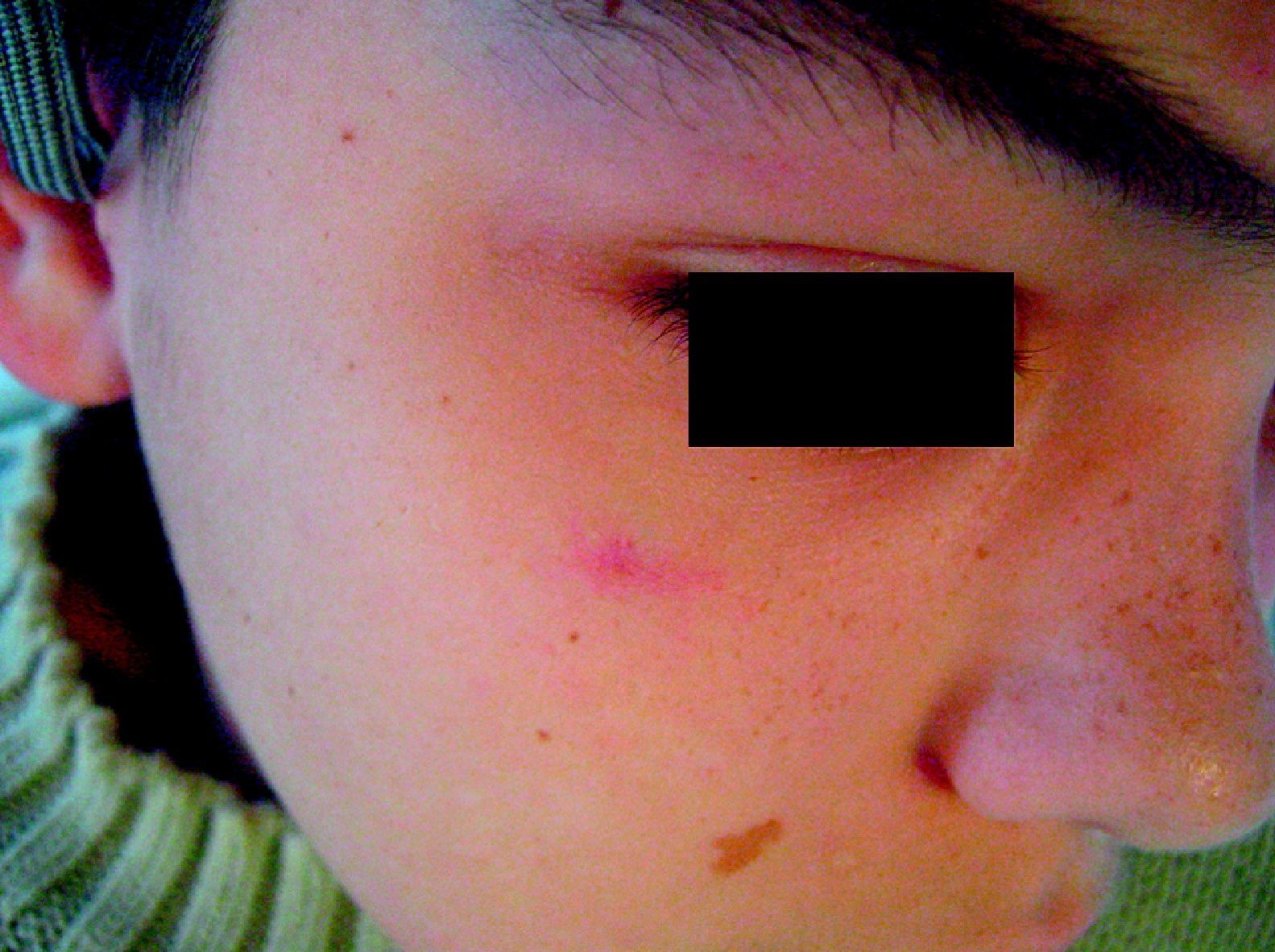
Nevus roseusEn el año 2005 Happle31 presentó una variante de las MVO o nevus flammeus caracterizada fundamentalmente por presentar una tonalidad rojo pálida, e incluso rosada, a diferencia de la tonalidad más o menos intensa que suelen tener las MVO (fig. 4). Por analogía con la MVO propone llamarla «mancha en vino rosado», pero en ocasiones en la infancia las MVO tienen una tonalidad rosada; por ello, a veces el diagnóstico de certeza no se hace hasta la edad adulta. La evolución natural de la MVO es hacia la hipertrofia, oscurecimiento y nodularidad, como se ha comentado; el nevus roseus permanece así toda la vida.
Puede plantear diagnóstico diferencial también con las manchas salmón, pero a diferencia de estas y al igual que las MVO, suelen ser lesiones lateralizadas, no mediales.
Uno de los factores que más apoya la individualización de esta entidad es su asociación con la facomatosis pigmento vascular de tipo iii o «spilorosea».
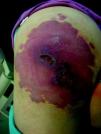
Cutis marmorata telangiectásico congénitoEs una malformación vascular congénita, rara y esporádica descrita por Van Louizen en 1922. Se presenta como lesiones eritemato-telangiectásicas de aspecto reticular que casi siempre aparecen en el nacimiento o en los primeros días de vida y pueden ser localizadas o tener distribución generalizada32 (fig. 5). En la mayor serie publicada de 85 pacientes33 los autores refieren que el 60% son localizadas y el 65% unilaterales, afectando estos casos con mayor frecuencia las extremidades inferiores. El 20% de estos pacientes presentaban MC adicionales en otra localización, como MVO, nevus anemicus o angioqueratomas34. Incluso se ha descrito un caso que comenzó como una MVO35. Es más frecuente la afectación de extremidades inferiores, y en un tercio de estos casos las lesiones de cutis marmorata telangiectásico congénito (CMTC) se acompañan de atrofia. En las extremidades superiores, sin embargo, es más frecuente que se acompañen de hipertrofia hasta en un 20% de los casos.
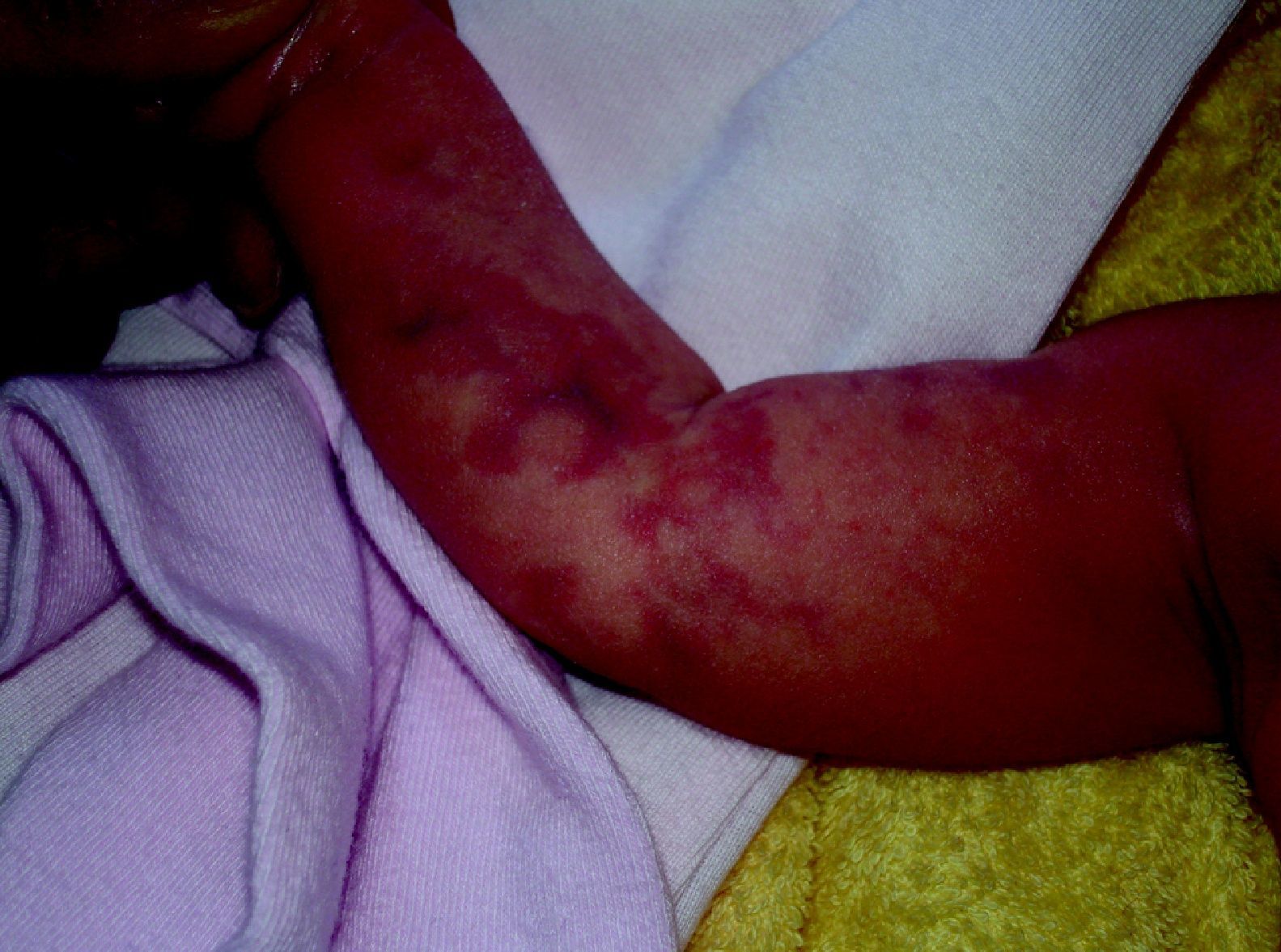
En un 20% de casos se asocian a otros procesos (tabla 3) como hemangiomas, nevus pigmentario congénito, mancha café con leche, hipospadias, glaucoma y malformación arteriovenosa hemisférica, sindactilia, aplasia cutis y malformación cardiaca, enfermedad renal multiquística e hipotiroidismo congénito36.
Cutis marmorata telangiectásico congénito. Asociaciones
| Hipotiroidismo congénito |
| Glaucoma |
| Hemangioma |
| Nevus pigmentario congénito |
| Máculas café con leche |
| Hipospadias |
| Sindactilia y aplasia cutis |
| Enfermedad quística renal |
| Nevus flammeus |
| Lupus neonatal |
| Monoatrofia |
| Ascitis transitoria y niveles elevados de gonadotrofina coriónica |
| Niveles de cobre elevados y aumento de elastólisis |
| Síndrome de Adams Oliver |
| Manchas mongólicas extensas: facomatosis cesio marmorata |
| Enanismo primordial osteodisplásico microcefálico de tipo ii |
| Urticaria crónica autoimmune |
| Hipoplasia de venas iliaca derecha y femorales |
Es importante señalar que el CMTC es una de las manifestaciones que pueden confundirse con el lupus neonatal37,38; por ello en estos pacientes, según la historia clínica, puede ser necesario realizar estudios para descartar esta posibilidad.
La evolución de la mayoría de estos pacientes es hacia la desaparición espontánea en meses o años, pero hay algunos casos que persisten toda la vida. La asociación más frecuente son las MC, por lo que en algunos casos estas podrían ser una extensión de la enfermedad. Así, algunos de estos casos persistentes son verdaderas MVO de aspecto reticular.
Según la nueva clasificación de las facomatosis pigmentovasculares forma parte de la facomatosis cesiomarmorata. En estos pacientes se asocian a melanocitosis dérmicas extensas39.
Hay una entidad inicialmente denominada macrocefalia-CMTC que posteriormente también se ha relacionado con otras MC y que se comentará en los síndromes asociados.
Facomatosis pigmentovascularesSe denomina facomatosis pigmentovascular a la asociación de un nevus vascular con un nevus pigmentario extenso40 (fig. 6). Aunque fue descrita con anterioridad este nombre se debe a Ota en 1947. Inicialmente se clasificaron según el tipo de anomalía pigmentaria asociada en 5 grupos41 (tabla 4), pero posteriormente Happle ha hecho una nueva clasificación más resumida42. El modelo genético que explica la aparición de estas lesiones es el doble moteado alélico o didimosis, mucho mejor conocido en animales y plantas, en el cual las células pierden la heterocigosidad genética.
Clasificación de las facomatosis pigmentovasculares
| Clasificación clásica | |
| Tipo I | |
| Nevus flammeus y nevus epidérmico lineal pigmentado | |
| Tipo II | |
| Nevus flammeus y melanocitosis dérmicas y/o nevus anémico | |
| Tipo III | |
| Nevus flammeus y nevus spilus y/o nevus anémico | |
| Tipo IV | |
| Nevus flammeus, melanocitosis dérmicas y nevus spilus y/o nevus anémico | |
| Tipo V | |
| Cutis marmorata telangiectásico congénito y melanocitosis dérmicas | |
| No clasificables | |
| Serán tipo A o B según tengan o no tengan participación sistémica | |
| Clasificación resumida de Happle | |
| Facomatosis cesioflamea | |
| Melanocitosis dérmicas y nevus flammeus | |
| Facomatosis spilorosea | |
| Nevus spilus y nevus telangiectásico de tipo pálido (nevus roseus) | |
| Facomatosis cesiomarmorata | |
| Melanocitosis dérmica y cutis marmorata telangiectásico congénito | |
| Facomatosis pigmentovasculares inclasificables | |
| Correspondencia entre ambas | |
| Cesioflamea | IIA, IIB |
| Spilorosea | IIIA, IIIB |
| Cesiomarmorata VA, VB | |
| No clasificable IA, IVB | |
| El tipo i de la primera clasificación se elimina | |
El diagnóstico de estas lesiones es fundamentalmente clínico y se ha asociado sobre todo la forma cesioflammea a múltiples anomalías que se exponen en la tabla 5. Se han descrito relacionadas en ocasiones con malformaciones vasculares complejas, como el síndrome de Klippel-Trenaunay43.
Asociaciones descritas de la facomatosis cesioflamea
| Lesiones cutáneas |
| Nevus anémico, manchas café con leche, vitíligo generalizado, alopecia triangular congénita |
| Anomalías vasculares |
| Síndrome de Sturge-Weber, Síndrome de Klippel-Trenaunay, granuloma piogénico, malformaciones venosas |
| Alteraciones neurológicas |
| Convulsiones, atrofia cortical, Arnold-Chiari tipo i, sordera bilateral, parálisis facial idiopática, hidrocefalia, diabetes insípida, neurofibroma plexiforme, retraso psicomotor |
| Alteraciones oculares |
| Melanosis ocular, mamilaciones y hamartomas del iris, glaucoma, vasos prominentes en la esclera, edema crónico de la córnea, alteraciones pigmentarias de la retina, catarata pigmentaria |
| Miscelánea |
| Discrepancia en longitud de extremidades, escoliosis, disrafismo espinal, hemihipertrofia, sindactilia, macrocefalia, agenesia renal, angiomatosis renal, hepatoesplenomegalia, hernia umbilical, hipoplasia de las venas de las piernas, déficit de IgA, síndrome hiper IgE, eccemas y erupción prematura de los dientes |
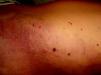
Las telangiectasias son vasos pequeños, dilatados de forma permanente. Aparecen en la piel como máculas rojizas puntiformes, como lesiones lineales o estrelladas y en muchos casos, en su evolución, sobre todo en la cara, se hacen papulosas (fig. 7).
Aunque desde un punto de vista teórico las causas de las telangiectasias en la infancia son muchas (tabla 6), en la práctica son poco frecuentes, y son pequeñas lesiones puntiformes idiopáticas, acompañando a síndromes congénitos o colagenopatías. Es muy frecuente que las arañas vasculares se localicen en los párpados inferiores.
Causas de telantiectasias en la infancia
| Idiopáticas o telangiectasia primaria de la infancia |
| Genodermatosis |
| Ataxia telangiectasia, síndrome de Bloom, síndrome de Rothmund-Thomson, enfermedad de Rendu Osler, enfermedad de Coats, esclerosis tuberosa, síndrome de Hallermann-Streiff, xeroderma pigmentoso, síndrome de Morquio, disqueratosis congénita |
| Conectivopatías |
| Lupus eritematoso, esclerodermia, síndrome de Raynaud |
| Otras dermatosis |
| Rosácea, papulosis atrófica maligna, telangiectasia esencial generalizada, telangiectasia nevoide unilateral, angioma serpiginoso |
| Metabólicas/hormonales |
| Síndrome carcinoide, síndrome de Cushing, diabetes, hipertiroidismo, hepatopatías |
| Tumorales |
| Lesiones residuales de hemangiomas, mastocitosis (telangiectasia macular eruptiva persistente), micosis fungoide, enfermedad de Hodgkin |
| Infecciosas |
| Tuberculosis, infección por el VIH |
| Miscelánea |
| Nevus aracneus o angioma estelar, enfermedad de injerto contra huésped, fármacos/tóxicos |
El tratamiento de estas lesiones se realiza mediante cualquier sistema de láser vascular específico con excelentes resultados44. Hay, sin embargo, un grupo de pacientes en que las telangiectasias son mucho más resistentes a los tratamientos con láser, como las telangiectasias residuales de los hemangiomas, la telangiectasia nevoide unilateral, la telangiectasia asociada a conectivopatías y las telangiectasias asociadas a la enfermedad de Rendu-Osler-Weber.
La telangiectasia nevoide unilateral tiene una forma congénita rara que predomina en varones, y una forma adquirida que afecta a niñas en época puberal. Suelen localizarse en la cara, el cuello, la parte superior del tórax y los miembros superiores45.
La ataxia telangiectasia o síndrome de Louis Barr es una enfermedad autosómica recesiva producida por la mutación del gen ataxia telangiectasia mutated (ATM) codificado en 11q22.3. Este síndrome, caracterizado por presentar telangiectasias que suelen empezar entre los 3 y 6 años, ataxia e infecciones recurrentes, asocia además alteraciones inmunológicas, por lo que estos pacientes suelen presentar neoplasias reticuloendoteliales. Un hecho bastante característico es la presencia de telangiectasias en la conjuntiva de forma bilateral46.
La telangiectasia hemorrágica hereditaria (THH), o enfermedad de Rendu-Osler-Weber, se caracteriza por telangiectasias en la piel y las mucosas, epistaxis recidivantes y su carácter familiar. Genéticamente heterogénea se transmite de modo autosómico dominante. En la THH tipo i está alterada la endoglina (parte del receptor del Tgf-beta, 9p33-34), en la de tipo 2 el gen ALK-1 (12q11-14), en la tipo 3 una mutación codificada en 5q31 y en la de tipo 4 en 7p14. Las lesiones cutáneas son poco frecuentes en la infancia y suelen aparecer tras la segunda década. En estas ocasiones también se asocian MAV en el sistema nervioso central, la médula, el hígado, el pulmón y el aparato digestivo (15-30% de los casos).
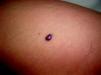
AngioqueratomasSon un grupo heterogéneo de lesiones que clínicamente se presentan como pápulas de rojo-violáceas a negras con superficie verrucosa (fig. 8) e histopatológicamente como dilataciones vasculares localizadas en la dermis papilar con hiperplasia epidérmica y marcada hiperqueratosis. Su terminología ha generado una enorme controversia en la literatura.
En la práctica clínica el principal problema que se plantea con el manejo de estos pacientes es que en lesiones que clínicamente son un angioqueratoma (AK) pueden subyacer diferentes malformaciones vasculares, y además existe una entidad, por el momento no clasificable, que probablemente deba estar aparte que es el hemangioma verrucoso. En la tabla 7 se hace una propuesta de clasificación de estas lesiones.
Clasificación de las lesiones tipo angioqueratoma
| Angioqueratomas clásicos (verdaderos angioqueratomas, malformaciones capilares) |
| Angioqueratomas de Mibelli: acrales asociados con frío |
| Angioqueratomas de Fordyce: en el área genital |
| Enfermedad de Anderson Fabry: angioqueratoma corporis difussum |
| Angioqueratoma corporis diffusum sin defectos enzimáticos |
| Angioqueratoma circunscrito: nevoide y comienza en edades tempranas |
| Angioqueratomas solitarios o múltiples que comienzan en la edad adulta |
| Lesiones tipo angioqueratomas como manifestación de otras malformaciones vasculares |
| Malformaciones capilares-linfáticas |
| Malformaciones venosas-linfáticas |
| Malformaciones capilares-linfáticas-venosas (síndrome de Klippel-Trenaunauy) |
| Malformaciones linfáticas |
| Malformaciones venosas |
| Las malformaciones cerebrales cavernosas causadas por mutaciones en KRIT1 se asocian en un 12% de los casos con manchas vasculares hiperqueratósicas que simulan un hemangioma verrucoso |
| Fístula arteriovenosa traumática |
| Hemangioma verrucoso |
Así, los 5 tipos clásicos de AK son las verdaderas MC. Los AK de Mibelli, los de Fordyce, el AK corporis diffusum, el circunscrito neviforme o Fabry de tipo ii y los AK adquiridos solitarios o múltiples.
El tratamiento electivo de estas lesiones es el láser, pero el de colorante suele necesitar varias sesiones para erradicarlas, ya que necesita sobrepasar la hiperqueratosis que presentan. Por este método suelen obtenerse resultados con menos sesiones con láser de CO2 y con láseres de Nd:YAG de pulso largo, los más utilizados en la actualidad.
Los AK de Mibelli son las primeras lesiones descritas a finales del siglo xix. Estas se relacionan con isquemia, y por tanto son características de zonas distales como los dedos de los pies o de las manos, las nalgas, las rodillas o los pabellones auriculares. La predisposición para padecer este tipo de AK parece transmitirse con una herencia autosómica dominante, con una penetrancia variable. Un subgrupo de estas lesiones se asocia a conectivopatías y hay algunos casos descritos en la infancia47,48.
Los AK de Fordyce son lesiones que están presentes en el escroto en el varón y en la vulva en la mujer. Se han relacionado con un aumento local de la presión venosa y son lesiones excepcionalmente vistas en la infancia.
El AK corporis diffusum (ACD) fue descrito por Anderson y Fabry a finales del siglo xix. Inicialmente relacionaron esta forma extensa de AK con una enfermedad metabólica, el déficit de alfa-galactosidasa A49. Posteriormente se han descrito 7 defectos enzimáticos (tabla 8) que pueden estar relacionados con la presencia de ACD, pero además hay casos de ACD que no se asocian a defectos enzimáticos, y casos de enfermedad de Fabry que no presentan ACD, sino unas pocas lesiones de AK o incluso sin lesiones cutáneas. Se ha descrito una paciente portadora con lesiones angiomatosas no angioqueratomas50. De hecho, la enfermedad de Fabry se diagnostica sobre todo en adultos, con la enfermedad totalmente desarrollada, por las manifestaciones renales o cardiacas51; desgraciadamente en la infancia solo se diagnostican los casos graves o los casos con antecedentes familiares conocidos. Una vez que se realiza el tratamiento sustitutivo de la enfermedad de Fabry en algunos casos desaparecen los AK52.
Defectos enzimáticos relacionados con el angioqueratoma corporis diffusum
| Alfa galactosidasa A: Anderson Fabry |
| Sialidasa: sialidosis tipo ii |
| Beta galactosidasa y neuraminidasa: galactosialidosis |
| Alfa fucosidasa: fucosidosis tipo ii |
| Alfa-N-aspartilgalactosaminidasa: aspartilglicosaminuria |
| o enfermedad de Kanzaki |
| Beta manosidasa: manosidosis |
| Gangliosidosis GM1 |
El AK circunscrito neviforme (ACN), también llamado enfermedad de Fabry de tipo ii, es raro en la infancia y suele aparecer a mediados o finales de la segunda década de la vida, siendo la localización más frecuente las nalgas53,54. Es más común en mujeres y se considera una forma segmentaria localizada de la enfermedad de Fabry55,56.
Por último, hay lesiones de tipo AK adquiridas de forma solitaria o múltiple que pueden presentar cualquier localización. Esta forma de presentación también es rara en la infancia.
Una lesión que hemos clasificado aparte, y que es típica de la infancia, es el hemangioma verrucoso57. Esta lesión que ha planteado gran controversia58,59, y de nombre es poco adecuado, se caracteriza por aparecer en el nacimiento y afectar sobre todo a las extremidades inferiores de forma segmentaria (fig. 9). Puede, sin embargo, presentarse como lesiones aisladas de pequeño tamaño en otras localizaciones. Suelen ser lesiones profundas afectando en ocasiones hasta el músculo, y con un componente hiperqueratósico muy marcado que las lleva a presentar episodios frecuentes de sangrado. Estas lesiones comparten algunos hallazgos de malformaciones y otros de tumores vasculares. Desde un punto de vista histopatológico tienen una característica peculiar, ya que más del 50% de los casos son GLUT-1+. Desde un punto de vista terapéutico si son lesiones de pequeño tamaño debe realizarse cirugía, y si son lesiones muy grandes hay que considerar el tratamiento con láser, aunque sea de forma paliativa.
Malformaciones venosas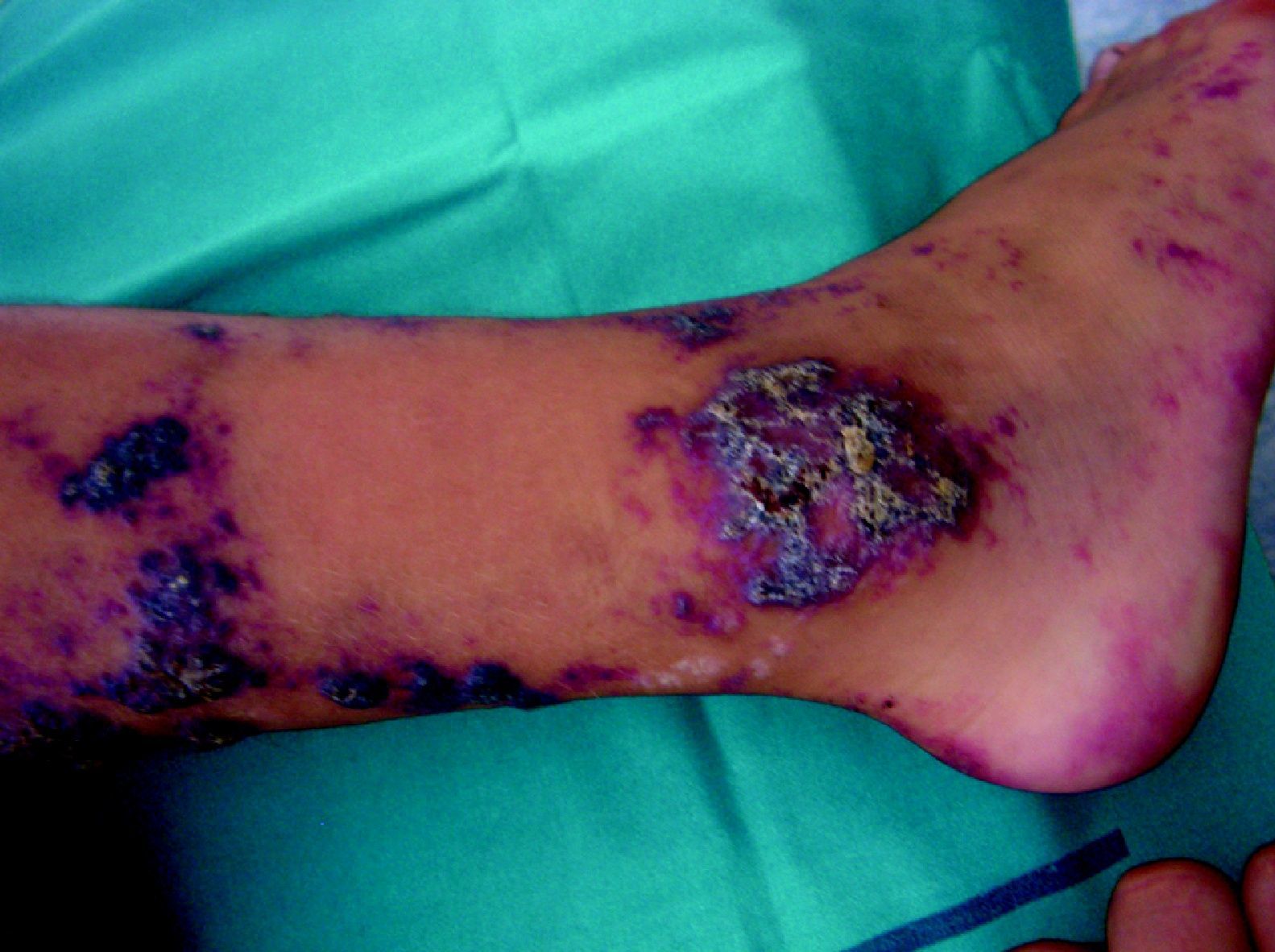
Las malformaciones venosas son malformaciones vasculares de flujo lento compuestas por canales venosos anómalos dilatados desde el nacimiento, con una expansión lenta durante su evolución. Presentan un mayor crecimiento hacia la pubertad y tras traumatismos.
En el pasado estas malformaciones han recibido nombres como angioma venoso, angioma cavernoso, hemangioma cavernoso, etc. Estos nombres deben ser abolidos, pues solamente conducen a confusión.
Para su mejor exposición las vamos a dividir en malformaciones venosas esporádicas, síndrome de Bean, malformaciones venosas cutáneo-mucosas familiares y malformaciones glomovenosas.
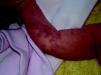
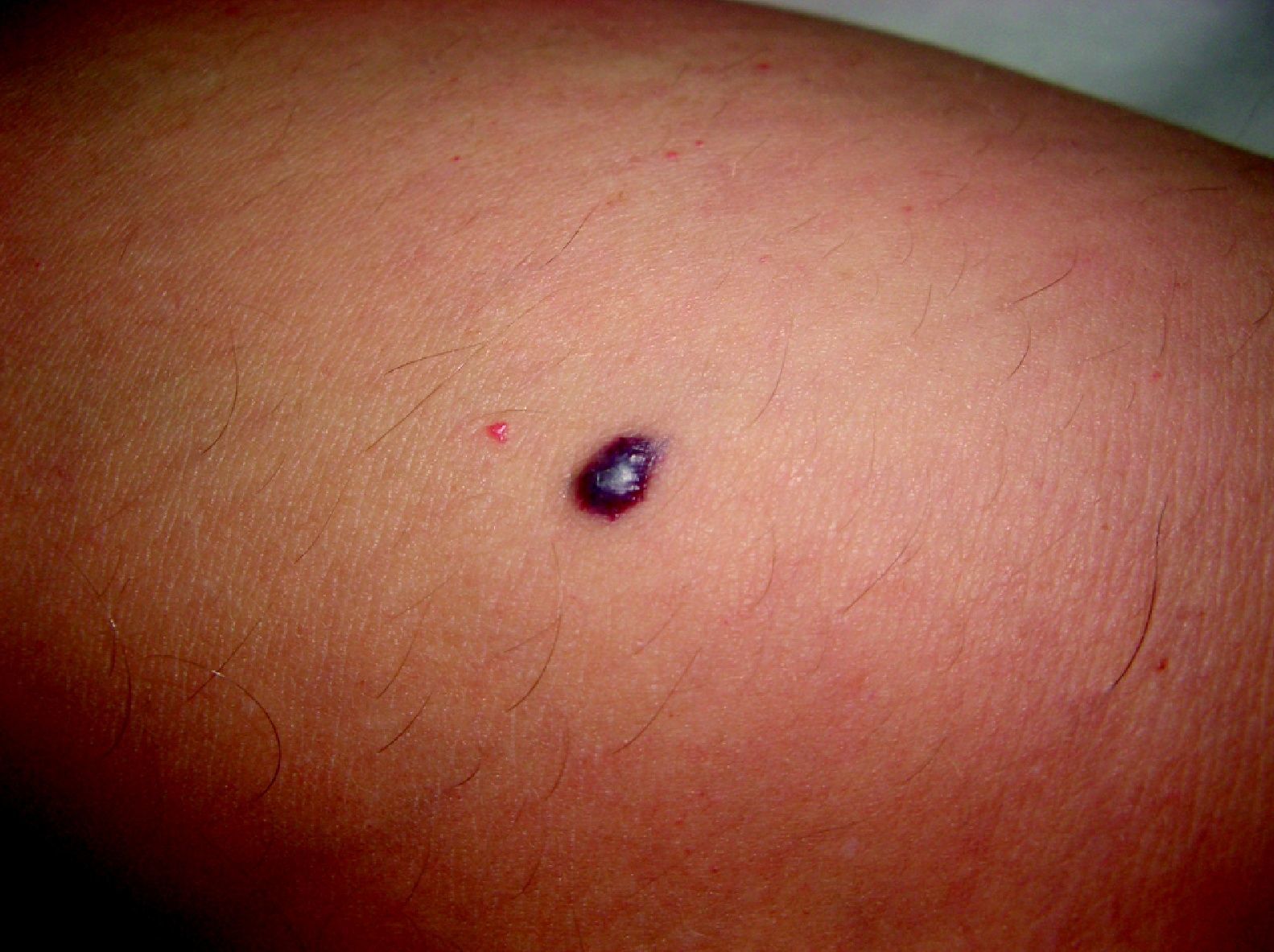
Malformaciones venosas esporádicasSe presentan clínicamente como masas azuladas o violáceas que se comprimen fácilmente, pero posteriormente se vuelven a rellenar (fig. 10). Es también característico que aumenten su tamaño con las maniobras que incrementan la presión venosa. Pueden ser únicas o múltiples, generalmente presentan bordes mal definidos, pueden ser localizadas o de gran extensión, superficiales o con afectación profunda, y aunque pueden presentarse en cualquier localización son más frecuentes en la cabeza y el cuello.
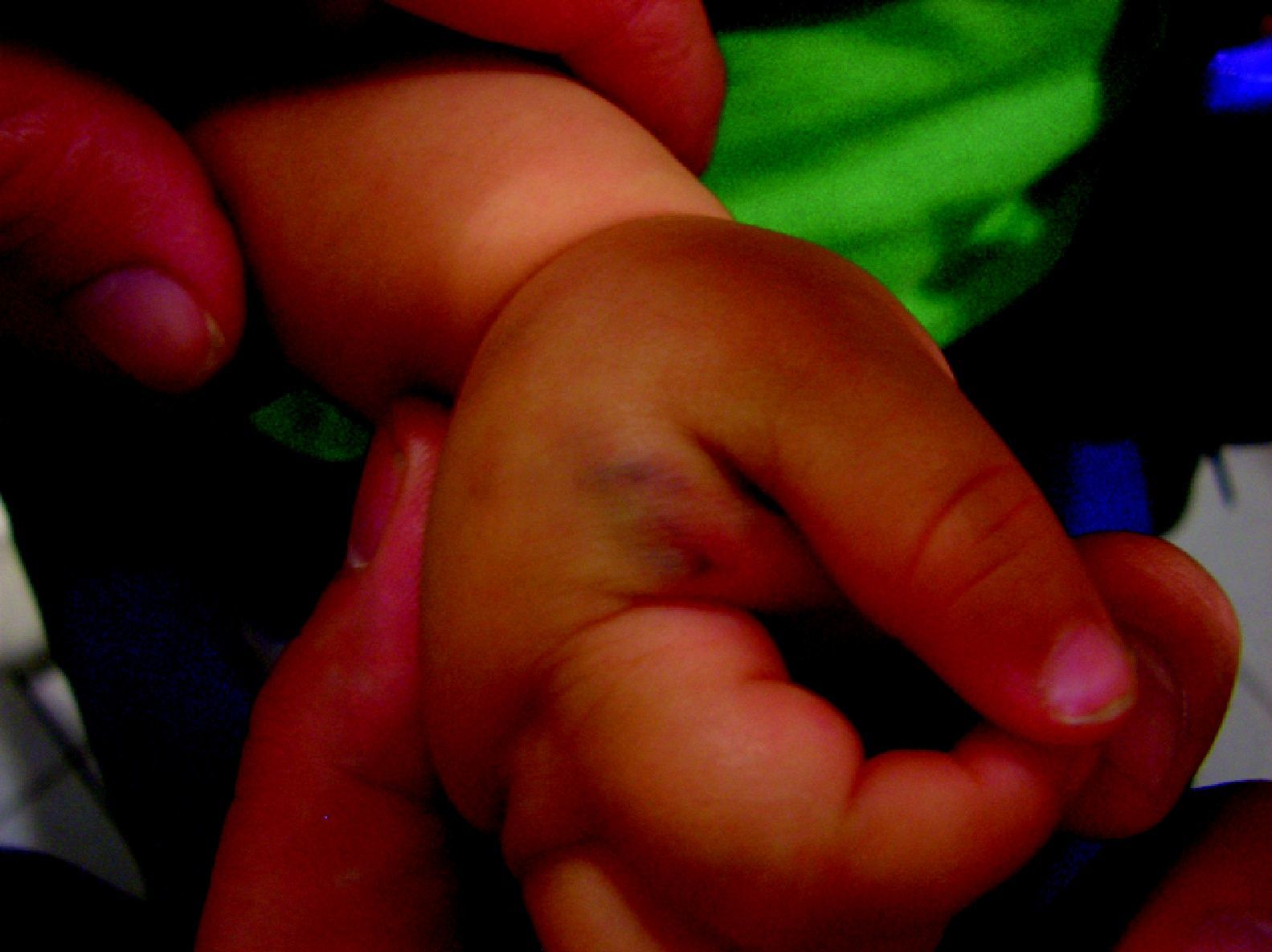
A la palpación no hay aumento de temperatura ni vibración (thrill). Los síntomas que suelen acompañarlas son dolor y edema de forma ocasional. En ocasiones estas manifestaciones aparecen de forma espontánea, generalmente por la presencia de fenómenos trombóticos en el seno de los vasos venosos tortuosos, y en otras tras traumatismos o cambios hormonales. Estos fenómenos trombóticos llevan a la formación de calcificaciones que pueden ser dolorosas a la palpación y que se denominan flebolitos. Son visibles con una radiografía simple y tienen un gran valor en cuanto al diagnóstico y al diagnóstico diferencial de estas lesiones.
Las lesiones profundas pueden invadir el músculo, las articulaciones y el hueso y llegar a producir deformaciones óseas y fracturas patológicas. Cuando afectan una extremidad entera es raro que induzcan una hemihipertrofia grave y progresiva, a no ser que formen parte de una malformación vascular compleja dentro del síndrome de Klippel-Trenaunay. La afectación articular puede inducir hemartrosis y por los fenómenos cicatrizales una artrosis precoz. La afectación muscular es de gran importancia, porque cuando afectan una zona inextensible como es un pie o una mano, el edema que se produce tras un traumatismo puede desencadenar un síndrome compartimental.
Estas lesiones pueden comenzar en la piel o en las mucosas, pero su presencia en la piel obliga a descartar la afectación profunda; de hecho, en ocasiones la parte visible de la malformación es solamente la punta del iceberg de la afectación profunda.
Por tanto, en estas lesiones es obligatorio realizar un estudio de imagen, que suele ser una angiorresonancia, para delimitar de la forma más precisa posible su extensión.
La evolución de estas lesiones es hacia un crecimiento lento pero progresivo que afecta en profundidad y en extensión superficial de las lesiones. Hay, sin embargo, algunos momentos en que pueden tener un crecimiento muy rápido, relacionado con una hemorragia intralesional, dentro de la malformación.
Sobre todo cuando estos pacientes tienen afectación muscular extensa se acompaña de una coagulopatía crónica que durante décadas se ha confundido con la trombopenia que acompaña al síndrome de Kassabach-Merritt60. Esta coagulopatía, que puede causar dolor agudo, esencialmente es una coagulación intravascular diseminada (CID) crónica que es muy importante conocer, pues en caso de cirugía por otros motivos puede producir problemas graves61,62. En la actualidad el principal marcador que se utiliza para controlar la evolución de esta coagulopatía son los niveles de dímero-D63, con niveles normales de fibrinógeno. Estos hechos, raros en la infancia, suelen presentarse en la edad adulta si la malformación no es tratada.
Recientemente se ha descrito que los pacientes con malformaciones venosas con alteraciones de coagulación con el tiempo pueden desarrollar hipertensión pulmonar, por la suelta repetida de pequeños trombos que de manera asintomática se van instalando en el pulmón64.
En 2008 Zietz et al.65 describen una variante dentro las MV esporádicas que llaman nevus venoso. Son lesiones solitarias, esporádicas, de distribución segmentaria causadas por mosaicismo genético.
El tratamiento de estas lesiones se basa en la compresión, la cirugía, el láser y la escleroembolización. El método ideal es la cirugía, pero esta se puede realizar en pocos casos de forma curativa, ya que las MV suelen ser infiltrativas y la cirugía sería muy mutilante66. De este modo lo que siempre debemos hacer, si podemos, es iniciar un tratamiento compresivo que retarda la progresión de las lesiones y mejora sus síntomas. La escleroembolización percutánea67,68, bajo control de técnicas de imagen, se puede realizar con alcoholes como el etanol, la bleomicina y el etoxiesclerol; recientemente se han descrito excelentes resultados con espuma de polidocanol. Precisa tratamientos repetidos con anestesia general y en ocasiones mejora las lesiones lo suficiente como para permitir una cirugía posterior. El tratamiento con láser debe reservarse para lesiones superficiales (láser de Nd:YAG)69 o como tratamiento paliativo de la parte superficial de la malformación (láser de CO2 o Nd:YAG). Recientemente se está utilizando en láser de diodo endovenoso para tratar estas lesiones de forma aislada o asociado a otros tratamientos como embolización70.
Síndrome de BeanTambién denominado «síndrome del nevus azul en tetina de goma». Estas lesiones de típico aspecto venoso tienen dos características fundamentales, son multifocales, aunque en ocasiones al nacimiento las podemos ver como grandes masas venosas únicas, frecuentemente en el polo cefálico, y se acompañan de afectación visceral sobre todo digestiva, por lo que es frecuente que estos pacientes presenten sangre franca u oculta en las heces de forma continuada71 y anemia ferropénica (fig. 11). Otras veces, sin embargo, presentan síntomas neurológicos72, a veces como primera manifestación, y hay casos asociados a lesiones venosas multifocales cerebrales, anomalías venosas cerebrales de desarrollo o sinus pericranii73.
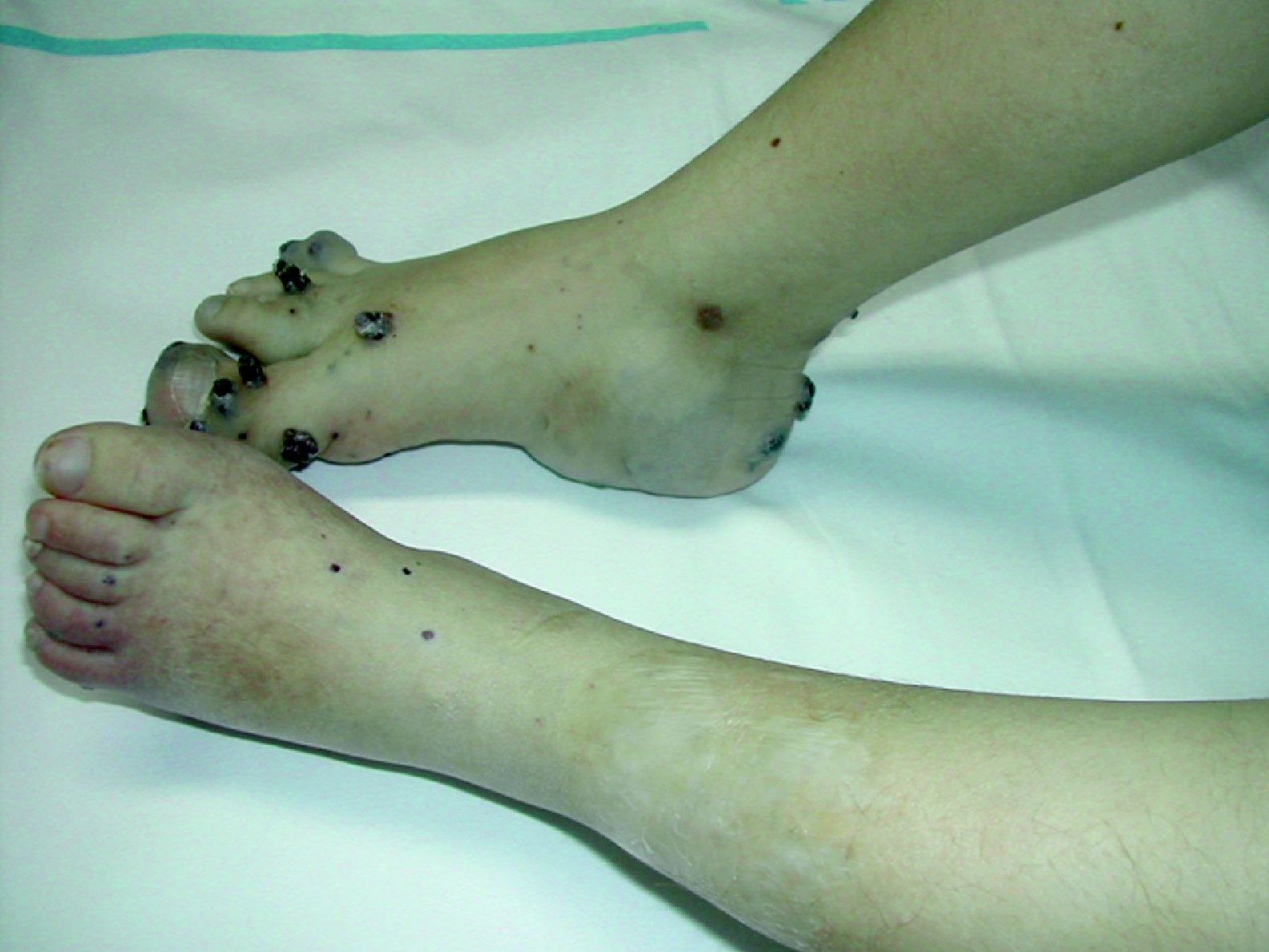
Es una entidad poco frecuente de etiología desconocida que normalmente se manifiesta de forma esporádica; cuando es hereditaria su modo de transmisión es autosómico dominante.
Las lesiones venosas en esta entidad van aumentando en número y tamaño de forma progresiva con la edad, y en los casos severos lleva a los pacientes a la muerte por sangrados gastrointestinales74. Recientemente se ha descrito el estudio de las lesiones intestinales en estos pacientes a través de una cápsula endoscópica75.
Normalmente en la infancia siempre están presentes, pero salvo en casos muy graves no suelen tener una sintomatología marcada76.
En algunos pacientes, al igual que en las MV, se puede producir una coagulopatía77 marcada por hipofibrinogenemia grave.
El diagnóstico diferencial con otras lesiones venosas, como las glomovenosas78, en ocasiones es difícil, pero la dermatoscopia79 puede sernos de ayuda, y el diagnóstico definitivo se realizará a través del estudio histopatológico.
El tratamiento de las lesiones viscerales es quirúrgico mediante enteroscopia y fulguración con arco de argón, y hay casos en que se realizaron decenas de resecciones término-terminales del intestino para poder eliminar totalmente estas lesiones y evitar las recidivas80,81. También se ha descrito el tratamiento con etidronato82 o con esclerosis con microespuma83, aunque solo es eficaz en lesiones aisladas.
Malformaciones venosas cutáneo-mucosas familiaresSe originan por una mutación del gen TIE-2/TEK codificado en el cromosoma 9p21-22 y el patrón de herencia es autosómico dominante84–86. El TIE-2 es un receptor de tipo tirosín-quinasa de las células endoteliales al que se liga la angiopietina-1. Tiene un papel muy importante en la vasculogénesis y en la angiogénesis, ya que los ratones deficitarios para TIE-2 desarrollan importantes alteraciones en la formación vascular87. El receptor TIE-2 alterado por una mutación produce activación independiente del ligando y presumiblemente suprime la apoptosis de las células endoteliales.
Siempre son lesiones cutáneo-mucosas clínicamente indistinguibles de las lesiones esporádicas. El resto de las consideraciones referidas a las esporádicas son aplicables a estas. De hecho, se ha demostrado que en las lesiones esporádicas también hay mutaciones del TIE-2, al igual que ocurre en las familiares86.
Malformaciones glomovenosasSon poco frecuentes y son malformaciones originadas a partir de las células glómicas endoteliales88. Antes todas estas lesiones eran conocidas como glomangiomas, pero en realidad son malformaciones y no lesiones tumorales.
Aunque pueden presentarse de forma esporádica, generalmente son familiares con un patrón de herencia paradominante, siendo la proteína alterada la glomulina codificada por un gen situado en 1p 21-22. A diferencia de las malformaciones venosas comentadas anteriormente, suelen presentar dolor a la compresión y no afectan las mucosas, el hueso, el músculo o las articulaciones, solamente la piel y el tejido celular subcutáneo. Un dato clínico que suele diferenciarlas fácilmente de las malformaciones venosas es que normalmente no son fácilmente depresibles, suelen tener un contenido más consistente y sólido.
Existen tres variantes clínicas, una forma solitaria que representa el 90% de las lesiones, cuya localización característica es el lecho ungueal, una forma múltiple generalmente familiar (que puede presentarse como lesiones extensas o de forma segmentaria) y una variante en placa de presentación atípica y que se ha descrito de forma congénita89.
El tratamiento de estas lesiones, en la mayoría de las ocaciones, es la cirugía, pero en los casos en que son pocas lesiones de pequeño tamaño el láser de Nd:YAG de pulso largo, que tiene una penetración de varios milímetros, constituye una gran opción terapéutica.
Por último, se ha descrito la asociación de malformaciones venosas cutáneo-mucosas con malformaciones venosas cerebrales familiares (carvernomas) relacionados con el gen KRIT190. Estos pacientes también presentan en ocasiones MC o lesiones de tipo AK en la piel.
Malformaciones linfáticasEl sistema vascular linfático es esencial para mantener el balance de líquidos, la repuesta inmune y la absorción de las grasas. Cuando se producen alteraciones en el sistema linfático desde la etapa embrionaria aparecen las malformaciones linfáticas91.
En estas malformaciones vasculares de flujo lento la terminología ha sido tremendamente confusa92.
Las malformaciones linfáticas que afectan a la piel se pueden clasificar en dos categorías: linfedema y malformaciones linfáticas congénitas. El linfedema puede ser primario como resultado de una hipoplasia de los troncos y los nódulos linfáticos, y secundario a otros procesos, por disfunción del sistema linfático. Las malformaciones linfáticas congénitas se presentan como malformaciones quísticas únicas o múltiples, superficiales o profundas. Estas lesiones quísticas se dividen en macroquísticas, previamente llamadas higromas quísticos, microquísticas o mixtas93. Los términos linfangioma, linfangioma circunscrito, linfangioma simple, hemangioma verrucoso y AK circunscrito se han utilizado para denominar malformaciones linfáticas microquísticas94. Por último existe un gran grupo sindrómico asociado a malformaciones linfáticas (tabla 9).
Síndromes asociados a malformaciones linfáticas
| Enfermedad de Gorham |
| Síndrome de Hennekam |
| Linfangiectasia intestinal |
| Síndrome de Klinefelter |
| Síndrome de Klippel-Trenaunay |
| Linfedema-distiquiasis |
| Linfedema-hipoparatiroidismo |
| Enfermedad de Meige (lymphedema tarda) |
| Enfermedad de Milroy |
| Neurofibromatosis |
| Enteropatía pierde proteínas (Protein-losing enteropathy) |
| Síndrome de Stewart-Treves |
| Síndrome triploide |
| Síndrome de Turner |
| Síndrome de las uñas amarillas |
| Síndrome de Noonan |
| Síndrome de Edwards |
| Enfermedad de Maffucci |
| Síndrome de Patau |
| Síndrome de Aagenaes |
| Síndrome de Prader-Willi |
| Linfangioleiomiomatosis |
| Linfangiectasia pulmonar |
| Síndrome de Bean |
| Síndrome Proteus |
| Síndrome de Adams Olliver |
| Microcefalia-linfedema-displasia corio-retininana |
| Síndrome Emberger |
| Síndrome CLOVES |
| Síndrome CLAPO |
La aproximación genética a las malformaciones linfáticas no ha sido posible. Ocurren esporádicamente y de haber causas genéticas son probablemente mutaciones somáticas, ya que si ocurriesen en células germinales serían letales95.
El término linfangiectasia hace referencia a una imagen clínica de lesiones pseudovesiculosas de contenido claro, aunque pueden tener contenido hemático cuando tienen comunicación con el sistema venoso. Puede relacionarse con malformaciones linfáticas o con alteraciones secundarias a estas.
Estas lesiones están siempre presentes al nacimiento, aunque pueden no manifestarse hasta años más tarde. Su crecimiento es paralelo al crecimiento del niño, y es frecuente que por infecciones o traumatismos presenten episodios de inflamación, en ocasiones de gran intensidad, que pueden causar importantes problemas, ya que la localización más frecuente de estas lesiones es la cabeza y el cuello.
La linfangiogénesis o formación de los vasos linfáticos, que ocurre tanto en el embrión como en la vida adulta, es un hecho poco conocido, pero parece que el VEGF-C96 y el TIE-197 desempeñan un papel importante; de hecho, ratones deficitarios en TIE-1 presentan importantes alteraciones del sistema linfático. Se han desarrollado en los últimos años marcadores de vasos linfáticos entre los que destacan el LYVE-198 y el PROX-199, que son expresados por linfáticos embrionarios; el VEGFR-3100 que inicialmente está en todos los vasos del embrión, pero que posteriormente se restringe a los vasos linfáticos, es esencial para el mantenimiento y crecimiento de la células linfáticas endoteliales; el VEGF-C y VEGF-D que son los principales ligandos del VEGFR-3 y VEGFR-2.
Malformaciones linfáticas macroquísticasCuando en el desarrollo embrionario del sistema linfático falla la conexión entre los grandes sacos linfáticos con el sistema venoso, se desarrollan grandes cavidades quísticas con crecimiento progresivo que se han llamado higromas quísticos101. Si finalmente se establece la comunicación con el sistema venoso desaparecen, pero si esto no ocurre forman las malformaciones linfáticas macroquísticas, que en muchas ocasiones llevan asociadas también lesiones microquísticas.
Normalmente ocurren en el cuello, en las axilas o en la cara lateral del tronco, y estos grandes quistes pueden ser únicos o múltiples y estar o no interconectados. La piel suprayacente suele tener un aspecto normal, y con su crecimiento forman masas que pueden llegar a ser de gran tamaño. Están presentes al nacimiento en el 60% de los casos y se diagnostican en el 85% de los casos antes de los 2 años.
Su complicación más peligrosa es la hemorragia, que hace que presenten un episodio agudo de gran inflamación y puede llevar a la obstrucción de la vía aérea o a alteraciones oculares importantes, según la localización de estas lesiones. En ocasiones estos episodios de inflamación ponen de manifiesto la malformación que hasta ese momento había pasado inadvertida102.
El diagnóstico suele ser fácil con un simple estudio ecográfico, pero cuando se presenta se debe descartar siempre la existencia de derrame pleural o de ascitis.
El tratamiento de estas lesiones inicialmente se debe realizar con escleroembolización percutánea, y en niños se han obtenido excelentes resultados con picibanil (OK-472)103, doxiciclina o bleomicina. En los casos en los que la esclerosis sea inefectiva la cirugía puede resolver el problema o minimizarlo. La reducción de masa linfática aberrante disminuye de forma proporcional los síntomas y las complicaciones104.
Malformaciones linfáticas microquísticasCuando en el desarrollo embrionario hay un fallo en la conexión entre los vasos linfáticos distales con los canales linfáticos se producen las malformaciones linfáticas microquísticas, que en la literatura actual siguen denominándose linfangioma circunscrito. Estas estructuras, por tanto, están excluidas de la circulación linfática general, y no suele presentarse nunca linfedema como manifestación.
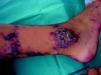
Estas lesiones tienen una presentación clínica mucho más variada. La forma más frecuente es como pequeñas pseudovesículas traslúcidas de contenido líquido claro que llamamos linfangiectasias y que pueden aparecer en cualquier localización, incluso en las mucosas (fig. 12). Estas, en ocasiones, por comunicación de la malformación con el sistema venoso con el que tienen un origen embriológico común, se llenan de sangre y presentan color violáceo. Estas vesículas suelen agruparse formando placas encima de la malformación subyacente, pero la extensión de la lesión cutánea no es un buen indicador de la parte profunda, que habrá que determinar con una prueba de imagen. Hay que tener en cuenta que en las lesiones linfáticas secundarias, como en el linfedema, en la parte superficial también pueden presentarse linfangiectasias.
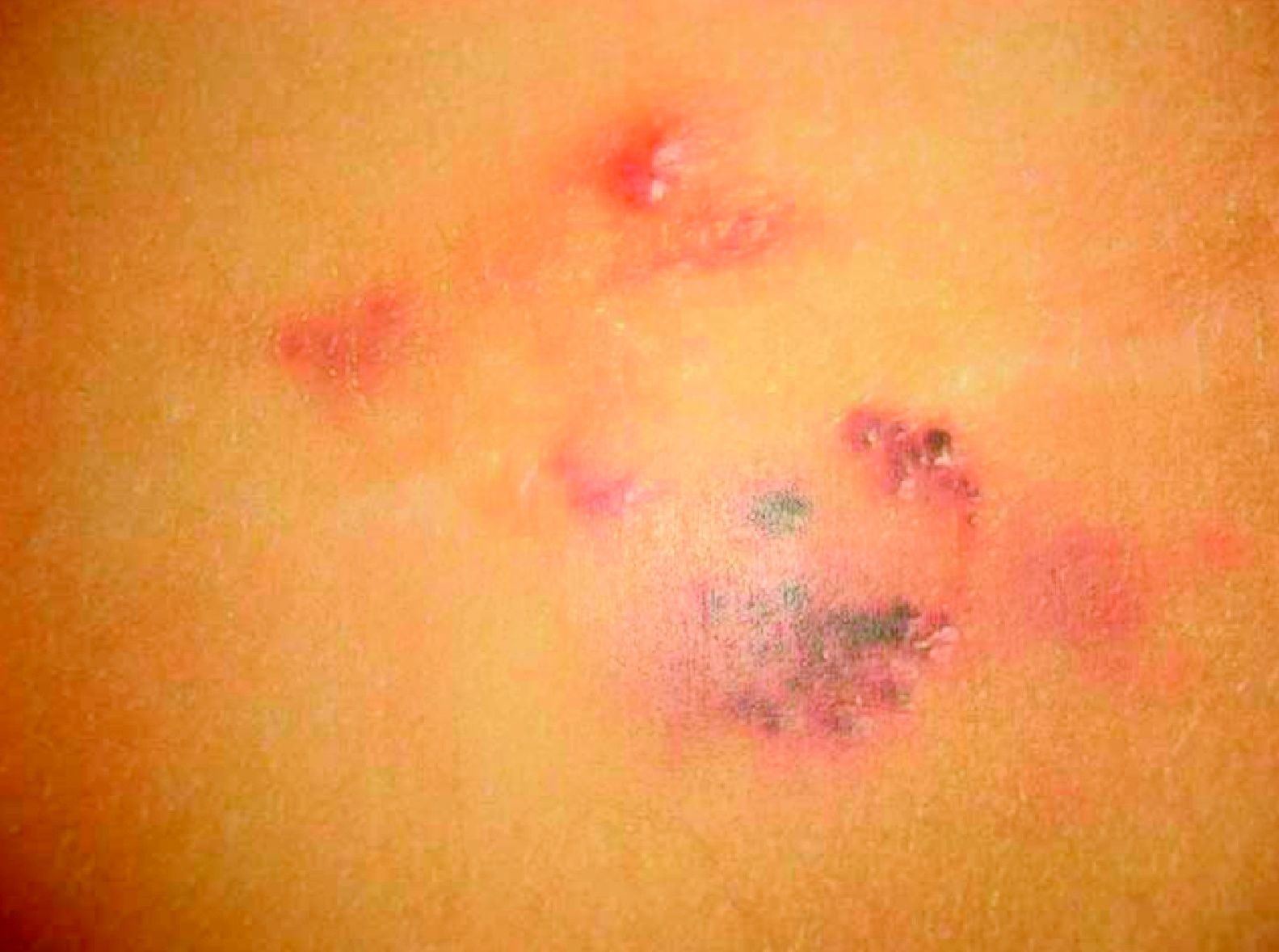
En nuestra experiencia la asociación de malformación capilar es signo de mal pronóstico, agresividad e infiltración por parte de la malformación.
Pero estas lesiones también pueden presentarse como lesiones verrucosas simulando verrugas o como lesiones verrucosas sobre un fondo violáceo, con aspecto de AK, de ahí el nombre de AK circunscrito que han recibido en ocasiones. Cuando estas lesiones son profundas en dermis papilar o en tejido celular subcutáneo podemos ver simplemente un poco de púrpura en la epidermis en los episodios de inflamación de modo que, en ocasiones, se ha confundido con maltrato infantil.
En el diagnóstico de estas lesiones la ecografía puede ser de gran ayuda, pero probablemente es más útil105 la realización de una resonancia magnética para determinar la extensión de las mismas106.
El tratamiento de estas lesiones es quirúrgico. Suele ser paliativo porque tienen una alta capacidad infiltrativa y la extirpación completa sin secuelas suele ser difícil, ya que excepcionalmente las pequeñas vesículas están comunicadas y permiten el paso de sustancia esclerosante entre ellas. En los casos localizados en la cavidad oral o en la lengua puede utilizarse láser de CO2107 o de Nd:YAG de pulso largo como tratamiento paliativo. También hay casos tratados con láser de colorante108. Recientemente se ha descrito el tratamiento de estas lesiones con un tratamiento combinado de radiofrecuencia y escleroterapia109.
Las complicaciones que pueden presentar estas lesiones se resumen en la tabla 10.
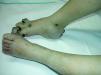
LinfedemaLa presencia de linfedema, la forma de patología linfática más frecuente en la edad adulta, nos debe orientar en la edad infantil a un defecto embriológico en la formación del sistema linfático, linfedemas primarios (fig. 13)110. El linfedema de Nonne–Milroy se caracteriza por la presencia de edema uni o bilateral de las piernas, brazos y/o cara con cambios fibróticos graduales e irreversibles (mutación en 5q34-35, gen que codifica el VEGFR-3111). El síndrome de Meige es un linfedema que aparece en la pubertad y suele acompañarse de episodios de celulitis112. Otro grupo de linfedema que aparece en la pubertad se describió asociado a distiquiasis (conjunto accesorio de pestañas), y es causado por haploinsuficiencia en el factor nuclear de transcripcción FOXC2113. El síndrome hipotricosis-telangiectasia-linfedema se ha asociado con el gen SOX18114.
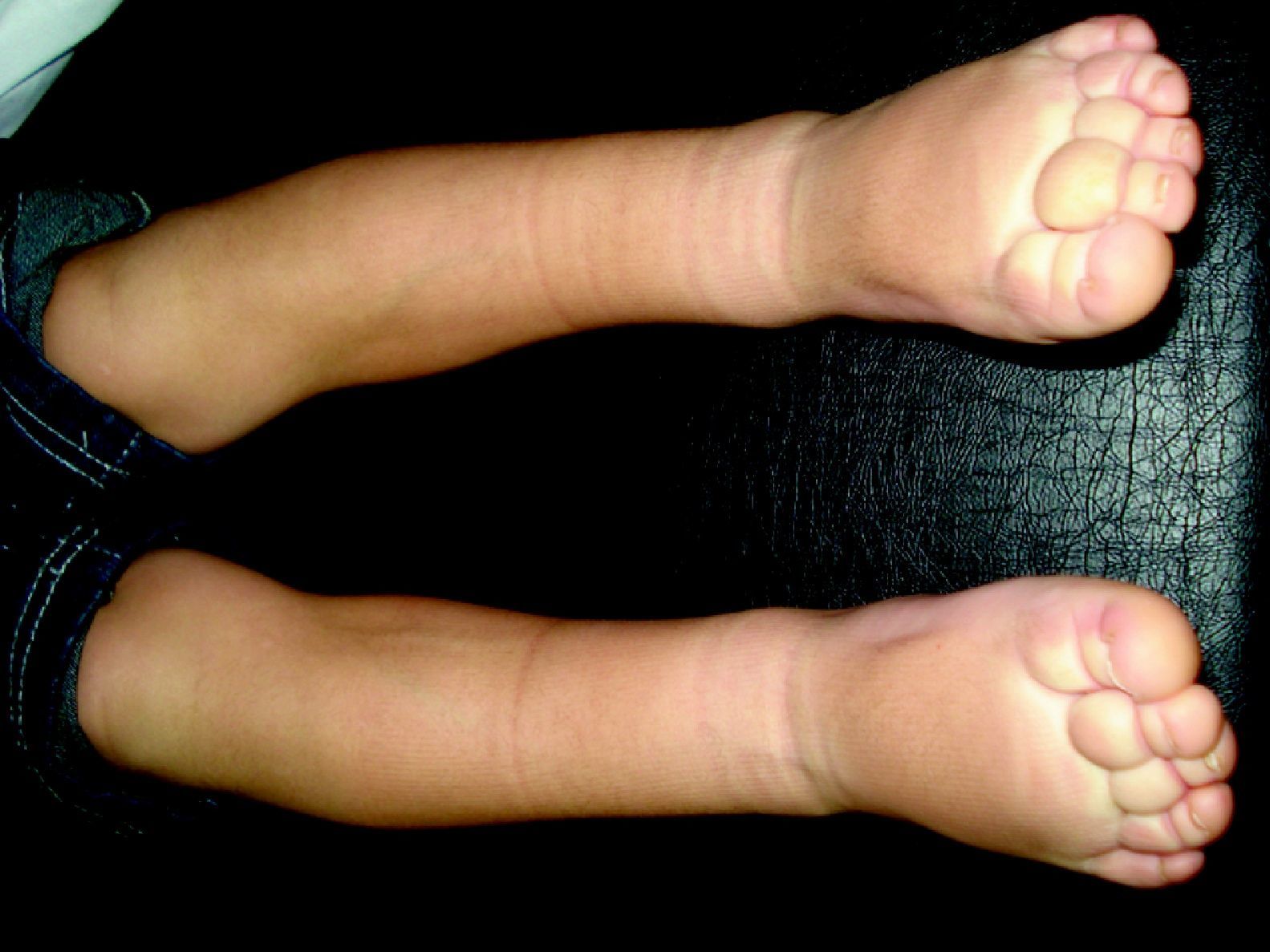
Otras formas de linfedema en la infancia son las asociadas a síndromes como el síndrome de Turner, el síndrome de Noonan o el síndrome de Klinefelter, que se describirán en la segunda parte de este trabajo.
Las formas secundarias de linfedema en la infancia aparecen generalmente como secuelas de infecciones cutáneas graves que han provocado linfadenitis, abscesificación y esclerosis de los linfáticos inguinales, con pérdida de su función.
Tratamientos de futuroEl desarrollo exponencial de la biología molecular en los últimos años hace que las expectativas en cuanto al tratamiento de las lesiones linfáticas sean muy importantes. Así, en animales de experimentación se ha demostrado que el tratamiento con VEGF-C recombinante induce crecimiento de la red capilar linfática y mejora los síntomas del linfedema secundario115,116. En este sentido también se ha demostrado que esta reestructuración linfática inducida por factores recombinantes incluye la regeneración del sistema valvular de los vasos linfáticos. El trasplante de nódulos linfáticos con VEGF-C restaura la anatomía de la red linfática en las áreas defectuosas, incluyendo los vasos y los nódulos linfáticos117.
Malformaciones de alto flujoEl alto flujo dentro de una malformación ocurre en lesiones puramente arteriales o en aquellas en que se produce una comunicación directa entre el sistema venoso y el arterial sin mediar el lecho capilar, es decir, el paso de un sistema de alta presión a otro de baja presión. Esto conlleva una circulación muy rápida del torrente sanguíneo que puede recogerse por un sistema de doppler, y cuando es más intenso lo captamos clínicamente como thrill o latidos.
Las malformaciones de alto flujo pueden ser malformaciones arteriales, fístulas arteriovenosas directas o MAV. Estas pueden presentarse de forma aislada o asociarse a una gran cantidad de síndromes (tabla 11).
Malformaciones arteriovenosas: síndromes asociados
| Síndrome de Bonnet-Dechaume-Blanc: MAV retina o coroides |
| Síndrome de Wyburn-Mason: sintomatología neurológica y alteraciones mentales |
| Síndrome de Brégeat: MAV conjuntiva, mejillas, nariz y boca |
| Síndrome de Cobb's: MAV medulares en el mismo metámero |
| Síndrome de Parkes Weber: MAV en un miembro |
| Enfermedad Rendu-Osler |
| Telangiectasia hemorrágica benigna |
| Enfermedad de Cowden |
| Neurofibromatosis |
| Hemangioma epitelioide |
| Síndrome Bluefarb-Stewart |
| Angiomatosis dérmica difusa |
| Síndrome de Lhermitte-Duclos |
| Acroangiodermatitis de Mali |
| Hiperplasia angiolinfoide con eosinofilia |
| Angioendoteliomatosis reactiva |
| Angioqueratoma corporis diffusum sin Fabry |
| Malformaciones capilares: alteraciones gen RASA-1 |
| Hamartoma folicular cutáneo múltiple |
| Tumores glómicos múltiples |
| Hemangiomas faciales |
| Aplasia cutis congénita |
| Síndrome de nevus sebáceo |
Las lesiones puramente arteriales como aneurismas, estenosis, aplasias o ectasias raramente se presentan en la piel. Las fístulas arteriovenosas directas, a diferencia del sistema nervioso central, también son raras en la piel y casi siempre ocurren como resultado de traumatismos. Por tanto, las lesiones que vamos a encontrarnos en la piel son las MAV localizadas o extensas.
Las MAV en la infancia son raras en comparación con las lesiones de bajo flujo. Las más frecuentes son las MAV cerebrales, que no son el objeto de este trabajo. Las MAV cutáneas, aunque siempre están presentes desde el nacimiento, es raro que den sintomatología de forma congénita o en los primeros años de vida. Suponen en la infancia un verdadero reto diagnóstico, pues generalmente son diagnosticadas de forma errónea hasta que están totalmente desarrolladas.
Schöbinger definió 4 estadios de progresión de estas lesiones. En el estadio i suelen ser máculas o parches rosados que simulan una malformación capilar anodina (fig. 14). Pero el aumento de temperatura o la vibración a la palpación sugieren un componente de alto flujo. En este estado suelen ser asintomáticas y permanecen de ese modo hasta la adolescencia, y algunas lesiones perduran así durante toda la vida. En el estadio ii la MAV se hace más prominente y los vasos más dilatados. La vibración y el latido son más intensos, se hacen patentes las venas de drenaje y en algunos casos la piel suprayacente se torna violácea. La progresión del estadio i al ii suele ocurrir en la pubertad en relación con cambios hormonales, traumatismos o tratamientos parciales de estas lesiones. El estadio iii se caracteriza por la destrucción de los tejidos profundos, incluido el hueso, y la presencia de dolor y hemorragia. El estadio iv se considera cuando el incremento del flujo induce una descompensación cardiaca.
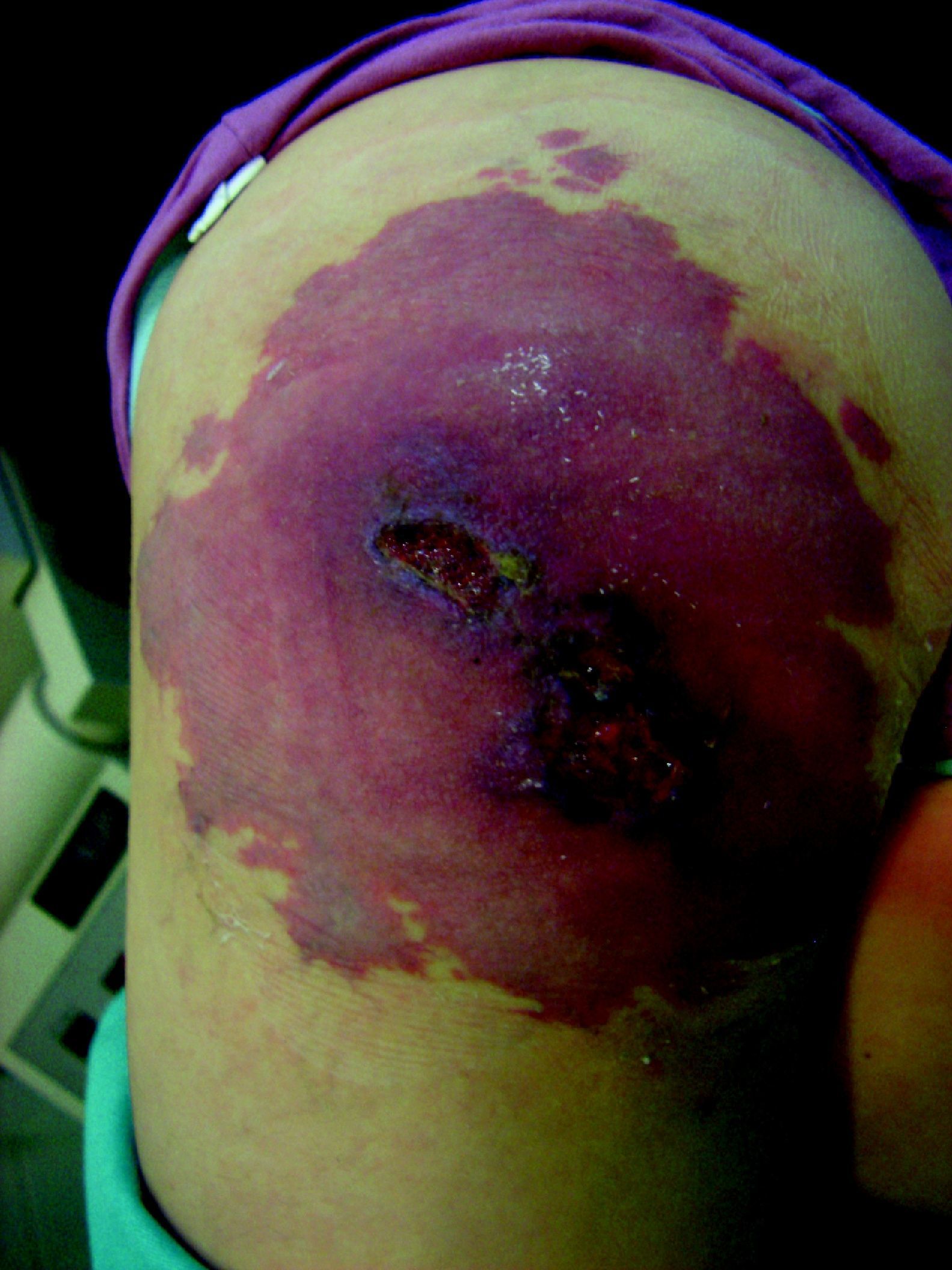
Sus manifestaciones clínicas son muy variadas (tabla 12), así como la sintomatología que pueden producir, limitaciones funcionales, dolor (más del 50% de los casos), hiperhidrosis, hipertricosis, hiperemia, vibración, cambios tróficos, ulceración, sangrado o lesiones de tipo pseudo-Kaposi118. Por ello, de forma general son las malformaciones vasculares de más difícil diagnóstico.
Malformaciones arteriovenosas: manifestaciones clínicas
| Mancha roja congénita |
| Venas dilatadas a menudo con thrill |
| Vibración cutánea |
| Lesiones pulsátiles |
| Una extremidad más larga desarrollada progresivamente hasta el final de la pubertad |
| Hipertrofia difusa con aumento de tamaño del miembro afecto |
| Hipertrofia localizada de los tejidos blandos |
| Linfedema y linfangiectasias |
| Pigmentación |
| Edema de pie |
| Fibrosis |
| Úlceras |
| Cambios cutáneos de pseudosarcoma de Kaposi (síndrome Stewart-Bluefarb) |
| Cuadros óseos líticos |
| Fallo cardiaco en presencia de FAV múltiples |
La eco-doppler tiene un papel determinante en el diagnóstico, así como la resonancia magnética, que delimitará la extensión de la lesión. Pero antes de realizar un tratamiento de estas lesiones es necesario hacer un estudio de imagen, angio-TAC preferentemente, que mostrará la morfología y la extensión de las conexiones arteriovenosas patológicas y que llamamos nidus.
Pero si su diagnóstico es difícil su manejo supone un verdadero reto que ha de afrontarse de forma multidisciplinaria, a pesar de lo cual muchos de estos casos no son llevados a buen fin. Las MAV tienen un comportamiento impredecible y una alta tasa de recurrencias, independientemente del tratamiento realizado119. El tratamiento de estas lesiones debe afrontarse realizando de forma inicial una embolización de la mayor parte de la malformación que sea posible y unos días después (24-48 horas) una cirugía para eliminar toda la lesión si es factible, o la mayor parte que conlleve la menor morbilidad. Es imprescindible eliminar el nidus de la lesión si queremos tener posibilidades de éxito. El hecho de realizar primero una embolización es que estas lesiones suelen englobar vasos de gran calibre, y así disminuimos de forma importante el sangrado en la cirugía.
El la infancia en pocos casos vamos a tener MAV con afectación cutánea en estadio ii de Schobinger o mayor, por lo que no es frecuente que tengamos que hacer tratamiento en esta edad.
Malformaciones vasculares complejasLa clasificación de las MV de la ISSVA, como se ha comentado, divide estas lesiones según el vaso predominante, pero en muchas ocasiones existe una predominancia de varios de ellos.
Las MC-linfáticas (MCL) suelen presentarse como una mancha en vino de Oporto, en el seno de la cual aparecen lesiones tipo AK que corresponden al componente linfático (fig. 15). También en ocasiones malformaciones linfáticas profundas en la cavidad abdominal presentan en la superficie una malformación capilar. Esencialmente la sospecha de una ML asociada a una MC debe hacer buscar afectación profunda con los estudios de imagen correspondientes.
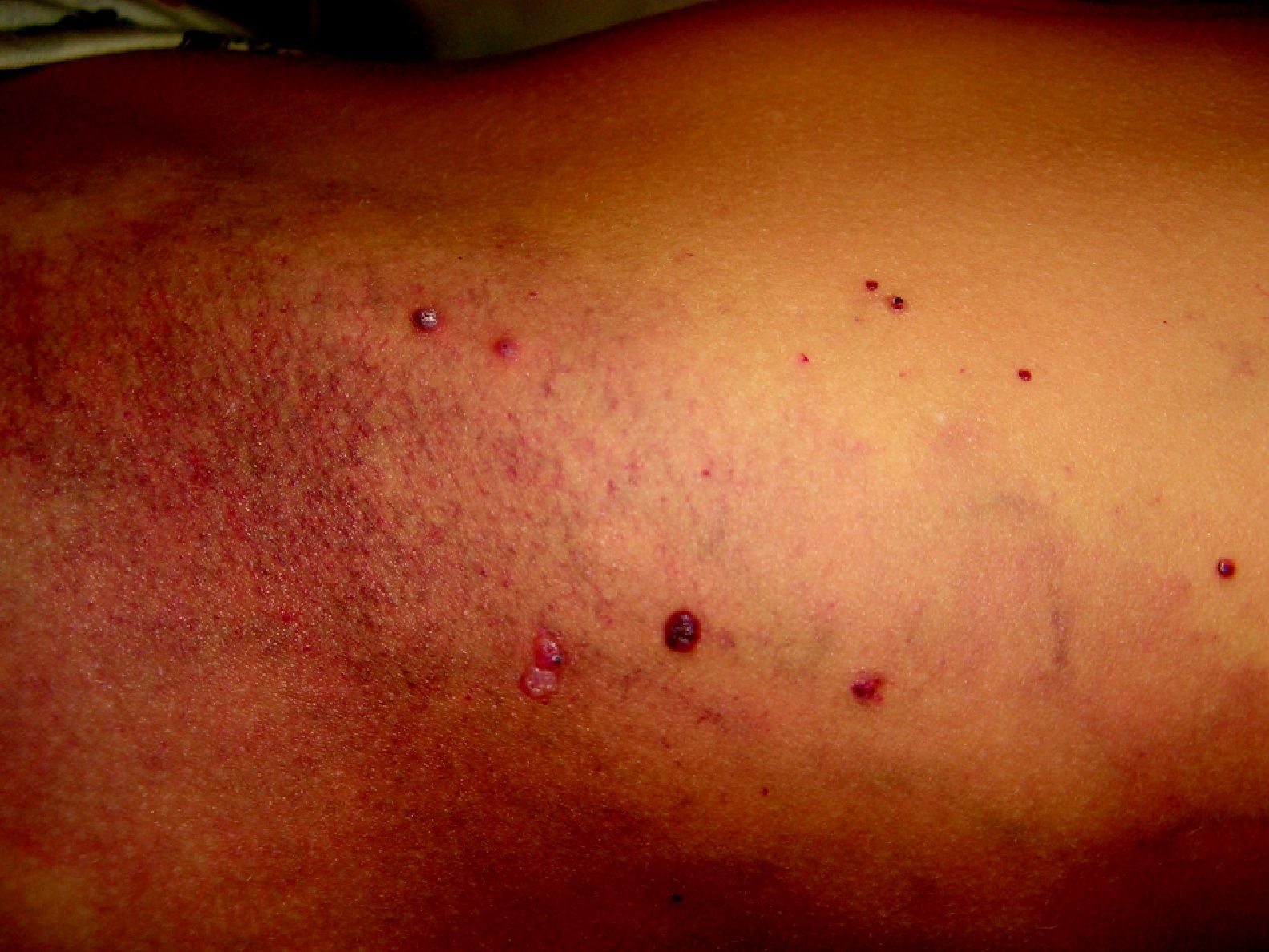
Las malformaciones capilares-venosas (MCV) se presentan como MVO, en el seno de las cuales vemos venas prominentes. Estas se presentan en muchas ocasiones en las extremidades inferiores sin acompañarse de una hipertrofia del miembro afecto.
Es importante diferenciarlas por tanto del síndrome del Klippel-Trenaunay (SKT), en el que debemos tener una malformación capilar, malformaciones venosas y linfáticas y una hipertrofia del lado afecto, porque el pronóstico es diferente. Hay casos muy raros de SKT en los que también se asocian MAV limitadas.
Las malformaciones veno-linfáticas tienen peor pronóstico de forma general que cualquiera de las dos formas aisladas. El tratamiento ideal en estos casos, siempre que sea posible, es la cirugía.
Las MC-venosas-linfáticas (MCVL) casi siempre las vemos en el contexto del SKT. En ellas la presencia de malformaciones linfáticas asociadas es un signo de mal pronóstico y de mala evolución.
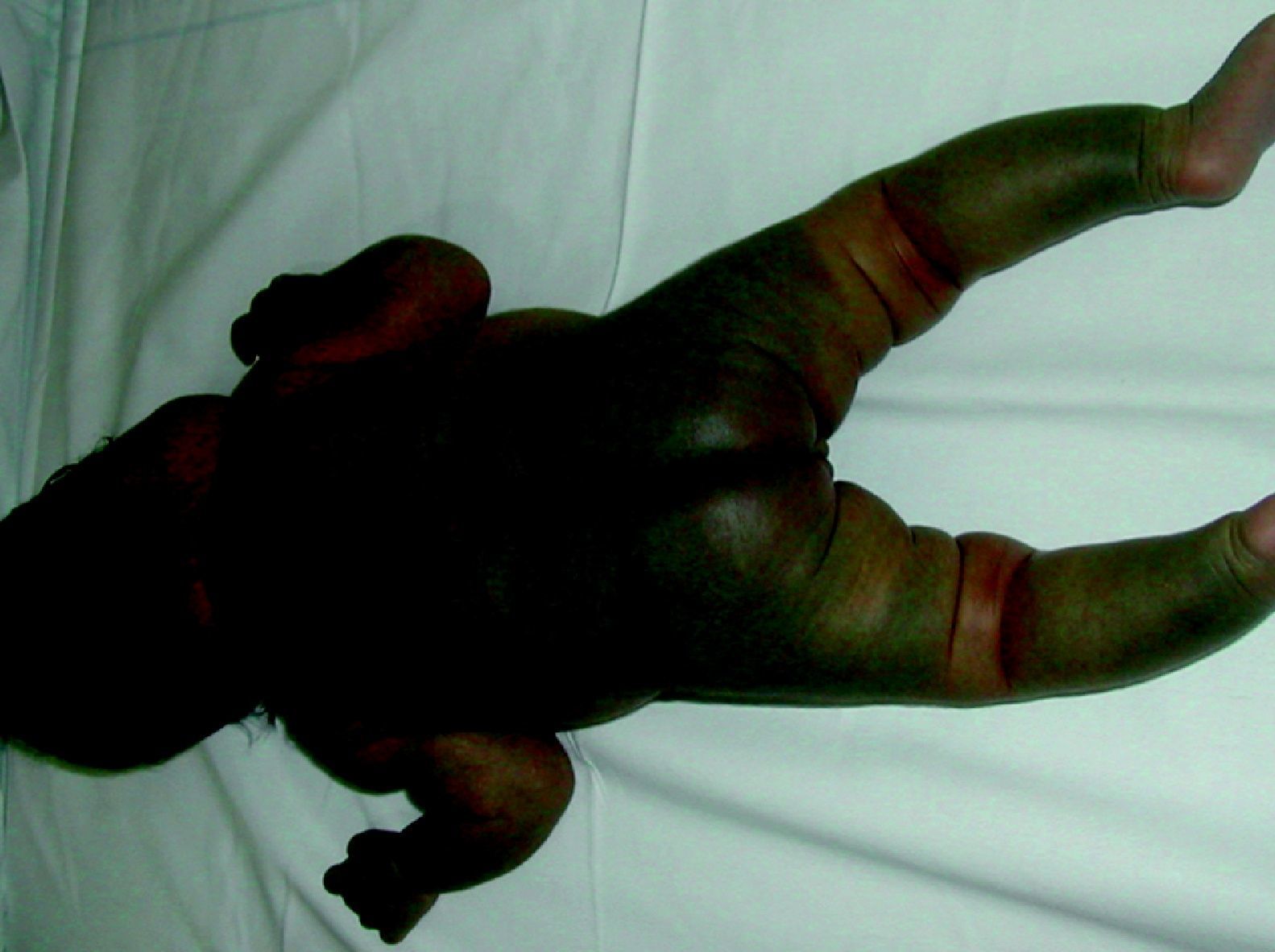
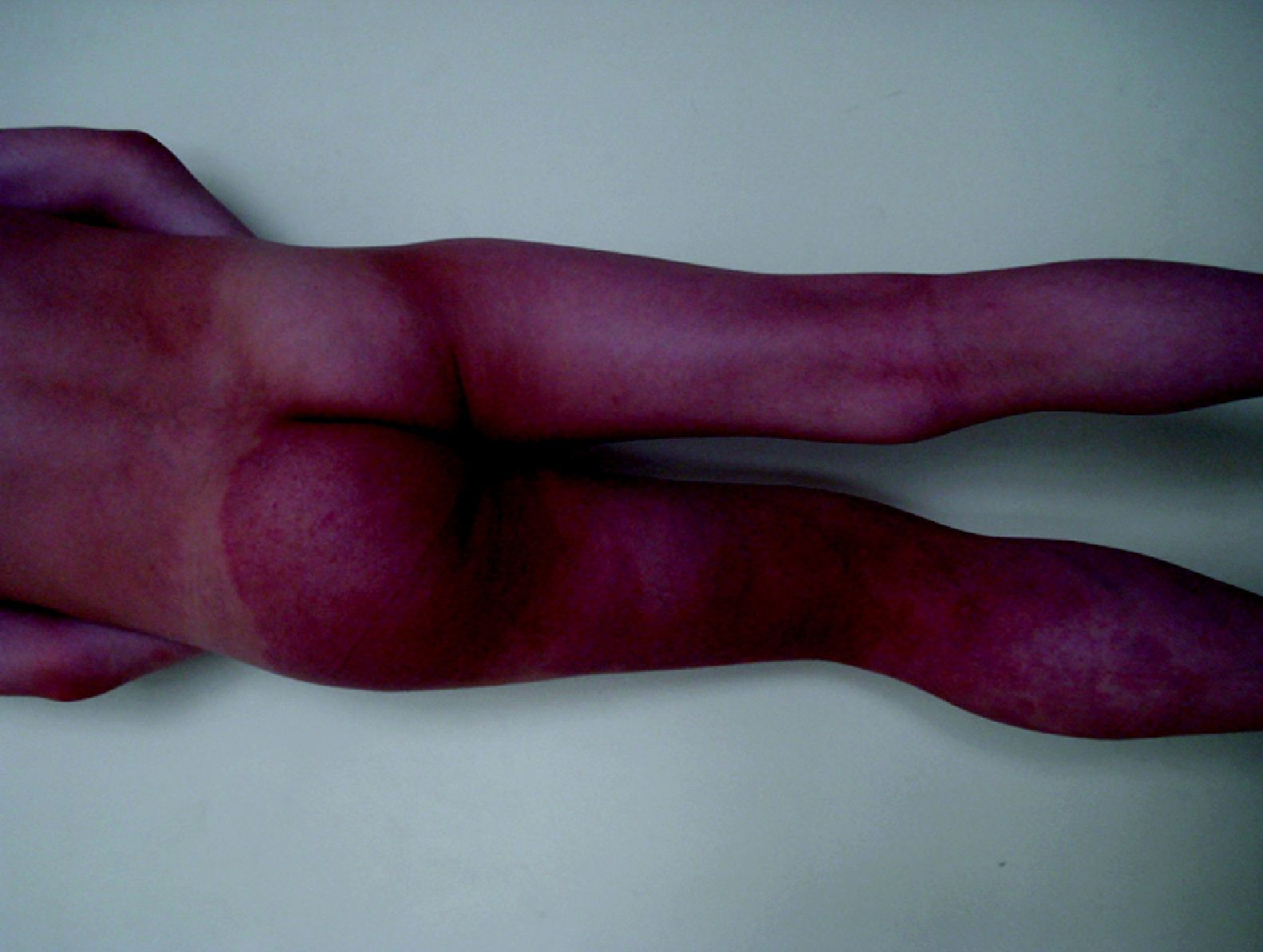
Las MC asociadas a MAV y a hemihipertrofia progresiva constituyen el síndrome de Parkes-Weber (fig. 16), de rara presentación y mal pronóstico. Hay también casos de MC múltiples, generalmente hereditarias, de presentación cutánea asociadas con MAV cerebrales, todo ello en relación con el gen RASA-1. También se ha demostrado positivo en muchos casos de síndrome de Parkes-Weber y hay muchos casos de MC familiares sin MAV que también son RASA-1 positivo.
La presencia de MC, venosas, arteriovenosas y linfáticas también se han descrito en el recientemente descrito síndrome CLOVES.
Malformaciones vasculares asociadas a tumores vascularesPara terminar, hay casos descritos en la edad pediátrica en los que se asocian malformaciones y tumores vasculares. Estos pueden ser hemangiomas congénitos y MC120, hemangioma infantil y cutis marmorata telangiectásico congénito, granuloma piogénico con MC121 o MAV122, hemangiomas segmentarios y anomalías arteriales (PHACE)123, hemangioendotelioma de células fusiformes y malformaciones venosas124, y hemangioendotelioma kaposiforme y malformación linfática125.
Por todo lo expuesto, y dada la complejidad que presentan estas lesiones y en muchos casos la poca frecuencia, deben abordarse desde un punto de vista multidisciplinario en centros de anomalías vasculares, como ya se hace en diversos centros de referencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


