



INTRODUCCIÓN
El linfoma subcutáneo de células T (LSCT) paniculítico es una rara variante de linfoma cutáneo de células T citotóxicas. Afecta preferentemente al tejido celular subcutáneo, y simula una paniculitis. Se ha incluido como una entidad clinicopatológica provisional por la Revised European American Classification of Lymphoid Neoplasms (REAL) en 1994 y posteriormente ha sido incorporada en la clasificación de la Organización Mundial de la Salud (OMS) como entidad propia1 .
No se tenía claro si esta clase de linfoma incluía una o varias características clinicopatológicas diferentes, pero a partir de la última reunión de la European Organization for Research and Treatment of Cancer (EORTC) en septiembre de 2004, se piensa que sólo hay un tipo bien definido. Todavía no se conoce cuál es el tratamiento más adecuado.
DESCRIPCIÓN DEL CASO
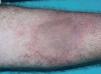
Un varón de 34 años, sin antecedentes de interés, presentó en julio de 2003 una lesión asintomática en cara anterior de muslo derecho, de crecimiento excéntrico y sin antecedentes de traumatismo o picadura. Un mes después, la lesión alcanzaba un gran tamaño, y se hizo dolorosa, asociándose a fiebre de 38 °C de predominio vespertino, escalofríos, sudoración profusa, pérdida de peso y astenia. Acudió a urgencias en varias ocasiones y fue tratado con doxiciclina (100 mg/12 h) durante 2 semanas, sin mejoría. En septiembre de 2003, con el diagnóstico de celulitis, se inició tratamiento con amoxicilina-ácido clavulánico (1 g/8 h), levofloxacino (500 mg/24 h) y antiinflamatorios no esteroideos. El paciente mejoró de su lesión cutánea y la fiebre se resolvió. Sin embargo, en octubre fue remitido a consultas de dermatología por el empeoramiento de la lesión cutánea. En la exploración física se observaba, en cara anterior de muslo derecho, una placa de unos 20 cm, indurada, con borde eritematoso activo, y centro marrón esclerodermiforme, con pérdida de vello que no fluctuaba ni crepitaba (fig. 1).
En las pruebas complementarias destacaba una leucopenia de 2.700/l (neutrófilos 1.200/l, linfocitos 1.000/l), con serie roja y plaquetas normales, así como una lactato deshidrogenasa de 638 U/l. Se realizó una biopsia cutánea, en la que se observó una paniculitis predominantemente lobulillar a expensas de un infiltrado mixto linfohistiocitario, y no se detectaron microorganismos con las técnicas de Giemsa, ácido peryódico de Schiff y Ziehl-Neelsen. Los cultivos de la biopsia cutánea para bacterias, hongos y micobacterias fueron negativos. Se realizó una segunda biopsia de una nueva lesión nodular eritematosa en cara anterior, de pierna izquierda aparecida 15 días después. En esta se observaba un infiltrado en el tejido celular subcutáneo, predominantemente lobulillar, compuesto por una población polimorfa que incluía principalmente linfocitos con atipia nuclear, núcleos hipercromáticos y contorno irregular, junto con células macrofágicas que contenían restos nucleares en sus citoplasmas. Se observaban abundantes figuras de mitosis y prominente cariorrexis (fig. 2). Llamaba la atención la disposición de los linfocitos atípicos que, junto con los restos nucleares, rodeaban a las vacuo-las lipídicas, formando un anillo alrededor de ellas (fig. 3). Las células linfocitarias neoplásicas expresaban CD43, CD3, CD8 y eran negativas para CD20, CD79 alfa, CD4, CD56 y CD30. El índice proliferativo era de alrededor del 25 %. El estudio molecular mediante reacción en cadena de la polimerasa de los genes del receptor de las células T no fue valorable. Los estudios de extensión fueron normales y en la médula ósea no se encontraron infiltraciones ni hemofagocitosis.
Con el diagnóstico de LSCT sin afectación de médula ósea se inició precozmente tratamiento quimioterápico con ciclofosfamida, adriamicina, vincristina y prednisona (CHOP). Tras ocho ciclos, el paciente presentaba buen estado general, la fiebre había remitido. No ha vuelto a tener nuevas lesiones de linfoma. La lesión inicial en cara anterior de muslo derecho remitió, dejando una atrofia residual, principalmente a expensas del tejido celular subcutáneo.
DISCUSIÓN
El LSCT es un linfoma no hodgkiniano de células T citotóxicas que afecta al tejido celular subcutáneo2,3 . Es una entidad poco frecuente con menos de 100 casos descritos, que representan menos del 1 % de todos los linfomas no hodgkinianos extraganglionares.
Normalmente, el LSCT afecta a adultos jóvenes entre 30 y 50 años, aunque se han descrito casos en ni-ños4,5 . Algo más de la mitad de los pacientes son mujeres. Clínicamente se manifiesta como nódulos o placas, únicos o múltiples, usualmente no ulcerados (80 %). Se localiza principalmente en miembros inferiores y con menor frecuencia en tronco, extremidades superiores y cara6-8 . Clínicamente puede simular un eritema nudoso o una paniculitis crónica inespecífica, celulitis e incluso puede manifestarse como una placa de alopecia en cuero cabelludo 9 . Las lesiones pueden regresar de forma espontánea o persistir. Hasta una tercera parte de los pacientes son diagnosticados erróneamente de paniculitis, lo que conlleva retrasos en el diagnóstico de hasta 14 años, a pesar de realizar múltiples biopsias cutáneas. Todo esto hace que se asocie a un peor pronóstico 10 . En algo menos de la mitad de los casos se asocia a síndrome constitucional. Se detecta citopenia en el 20 % y un aumento de transaminasas en el 10 %. Las adenopatías suelen aparecer en estadios finales. Lo mismo ocurre con la médula ósea que, si se encuentra afectada, generalmente por hemofagocitosis, implica un mal pronóstico con curso mortal 11 . Se ha descrito transformación leucémica del LSCT en un caso 12 .
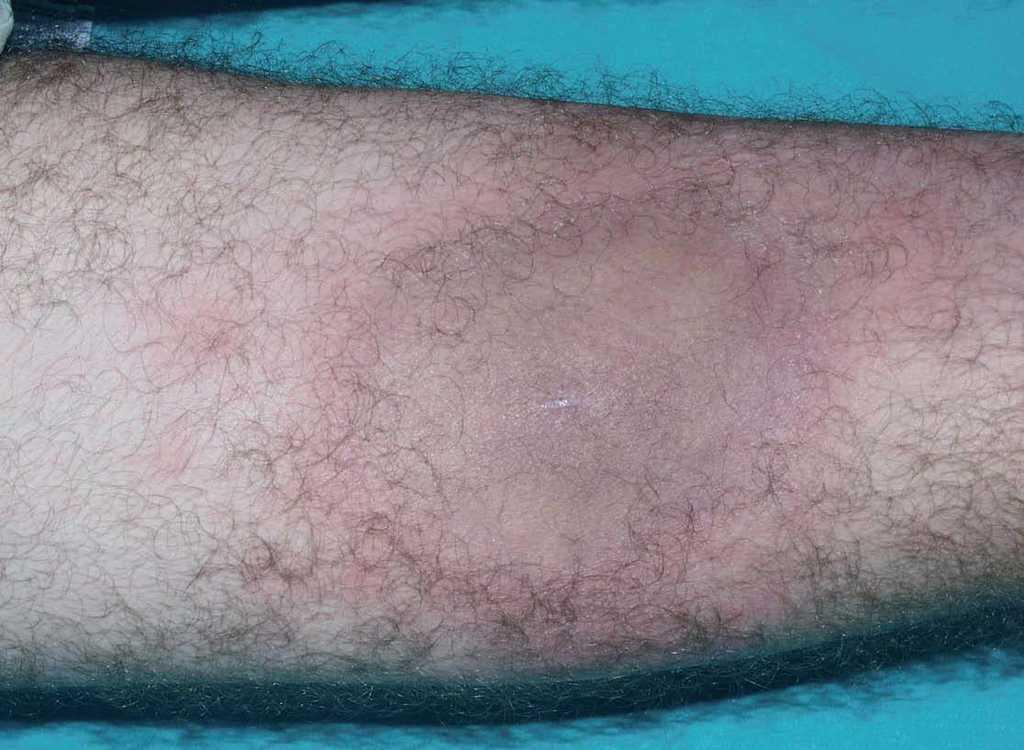
Fig. 1.—Placa indurada de unos 20 cm en cara anterior de muslo derecho.
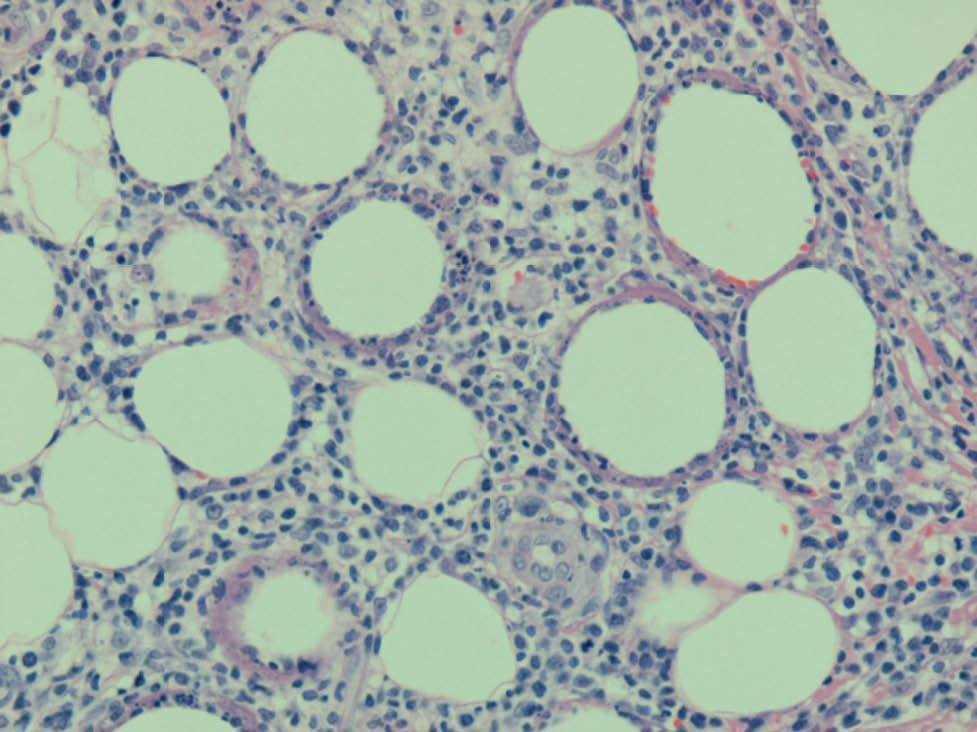
Fig. 2.—Infiltrado del tejido celular subcutáneo, predominantemente lobulillar, compuesto por una población polimorfa que incluye principalmente linfocitos atípicos, con núcleos hipercromáticos y contorno irregular, junto con células macrofágicas que contienen restos nucleares en sus citoplasmas. Se observan abundantes figuras de mitosis y prominente cariorrexis. (Hematoxilina-eosina, ×100.)
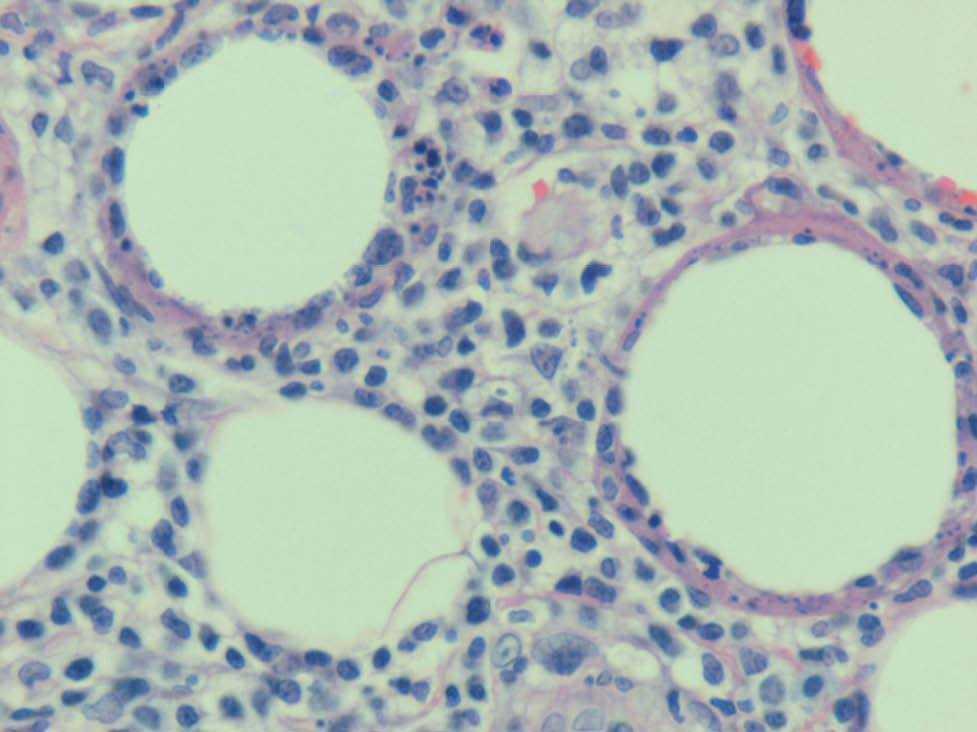
Fig. 3.—Llama la atención la disposición de los linfocitos atípicos que, junto con los restos nucleares, rodean a las adipocitos, formando un anillo alrededor de ellos. (Hematoxilina-eosina, ×200.)
El diagnóstico del LSCT es histológico. Puede ser difícil porque, en los estadios iniciales, no hay atipia y en las biopsias de larga evolución puede simular una paniculitis en resolución. Histológicamente en el LSCT se observa un infiltrado inflamatorio que afecta principalmente a los lobulillos grasos, compuesto por linfocitos T que muestran una marcada atipia, con núcleos grandes y cariorrexis. Es característico encontrar los linfocitos atípicos alrededor de los adipocitos necróticos formando un anillo. La mayor parte de los linfocitos atípicos expresan CD3, TIA-1 y CD8; ocasionalmente expresan CD4 y en pocos casos existe coexpresión CD8/CD4 o son ambos marcadores negativos. En el 50 % de los casos de LSCT en los que se han realizado estudios genotípicos se ha observado reordenamiento monoclonal del receptor de las células T6,7,13 .
Desde el punto de vista de la evolución el LSCT se clasificaba en dos subtipos diferentes. Un LSCT de curso indolente (82 %), parecido a una paniculitis histiocítica citofágica, con fenotipo de células T citotóxicas /, CD8+, CD56–, y supervivencia 5-10 años, y un LSCT de curso agresivo (18 %), fenotipo T citotóxico /, CD8–, CD56+, EBV+, con supervivencia de un año11,14,15 . Sin embargo, desde la última reunión de la EORTC en 2004, se considera sólo LSCT aquellos casos con fenotipo /, habitualmente CD3+, CD4–, CD8+, CD30–, CD56–, restringidos al tejido celular subcutáneo, en los que el síndrome hemofagocítico es poco frecuente y que, en general, presentan un curso indolente, con supervivencia a los 5 años del 80 %. El segundo subtipo de LSCT se engloba ahora dentro de un grupo aparte denominado linfomas T cutáneos /.
La localización subcutánea del infiltrado por sí misma no es una característica distintiva de este subtipo de linfoma. Puede observarse una sintomatología cutánea exactamente igual a la del LSCT en el grupo de linfomas T cutáneos / y en otros, como el linfoma de células natural killer /T (NK/T), linfoma T CD30+, linfoma de células B y micosis fungoide, entre otros16-18 . Entre ellos el diagnóstico diferencial debe realizarse principalmente con el linfoma NK/T y con el /. Son difíciles de distinguir de los LSCT porque a veces se superponen en las manifestaciones clínicas, en la histología y en las características inmunofenotípicas19,20 . Por otro lado, muchos pacientes que han sido diagnosticados de paniculitis histiocítica citofágica, son en realidad, verdaderos LSCT, ya que estas patologías se solapan21 . La paniculitis histiocítica citofágica es una verdadera paniculitis en la que se observa un infiltrado lobulillar de histiocitos y linfocitos T maduros junto a fenómenos de citofagocitosis, que se manifiestan como macrófagos «en bolsas de judías» (beanbags) . Según los diferentes autores, la paniculitis histiocítica citofágica podría ser un síndrome preneoplásico o un proceso reactivo a una enfermedad neoplásica, o bien que se trate de un linfoma de bajo grado al inicio y que puede evolucionar a un LSCT22-24 .
El tratamiento del LSCT es controvertido. En la serie de Weening et al3 , la mayor parte de los casos fueron tratados con quimioterapia obteniéndose remisiones completas en la mitad de los pacientes. Otros tratamientos realizados fueron radioterapia aislada o asociada a quimioterapia, con mejores respuestas, pero la diferencia no fue significativa por ser pequeño el número de pacientes. En los últimos años parece que los mejores resultados se han obtenido con tratamiento quimioterápico agresivo seguido de trasplante autólogo o alogénico de médula ósea25,26 . Este último está indicado en los casos con fenotipo agresivo y fenómeno de hemofagocitosis, actualmente incluidos en el grupo de linfomas T cutáneos /. La ciclosporina A también se ha propuesto como tratamiento coadyuvante en estos últimos casos con afectación de la médula ósea, con buenos resultados27 . El tratamiento de las formas menos agresivas debe ser quimioterápico, normalmente CHOP o variaciones del CHOP combinado o no con radioterapia28 .
Correspondencia:
María Rodríguez-Vázquez. Departamento de Dermatología. Hospital del Carmen. Ronda del Carmen, s/n. 13002 Ciudad Real. España. mrodvaz@yahoo.es
Recibido el 16 de julio de 2004. Aceptado el 27 de diciembre de 2004.


