



HISTORIA CLINICA
Un varón de 63 años acudió a urgencias por presentar una úlcera en la zona escapular izquierda. Como antecedentes patológicos destacaban un síndrome mielodisplásico diagnosticado hacía 4 años y que no requería tratamiento, diabetes mellitus no insulinodependiente, hiperuricemia y osteoporosis.
La lesión había aparecido unos 5 o 6 días antes como un nódulo que se ulceró, sin otra sintomatología acompañante.
Refería episodios previos, en años anteriores, de lesiones similares en la misma localización y en la escápula contralateral, que asociaban fiebre y curaban dejando cicatriz.
EXPLORACION FISICA
En la escápula izquierda se observó una úlcera ovalada, con bordes sobreelevados, eritematosos e infiltrados al tacto, y una escara necrótica central de 3 * 4 cm de diámetro (fig. 1).
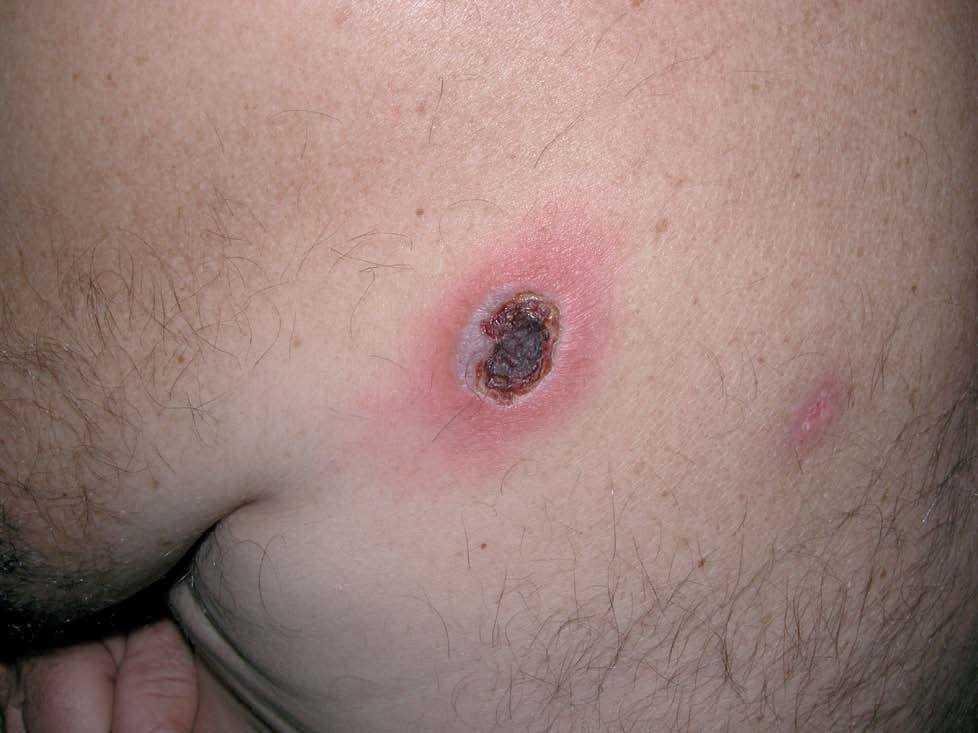
Fig 1.--Lesión ulceronecrótica en escápula izquierda.
En ambas escápulas se observaron lesiones cicatriciales redondeadas, algo deprimidas e hipopigmentadas (fig. 2).

Fig 2.--Lesión cicatricial en escápula derecha.
El resto de la exploración física no reveló ninguna alteración significativa.
EXAMENES COMPLEMENTARIOS
Se realizó biopsia en sacabocados del borde de la zona ulcerada de la lesión (fig. 3). La analítica realizada mostraba elevación de la velocidad de sedimentación globular (VSG) y leucopenia de 4.400/mm 3 con alteración de la fórmula leucocitaria (20,1 % neutrófilos, 54,9 % linfocitos, 23,3 % monocitos, 1,2 % eosinófilos, 0,5 % basófilos). El resto de los parámetros, así como la bioquímica se encontraban dentro de límites de la normalidad.
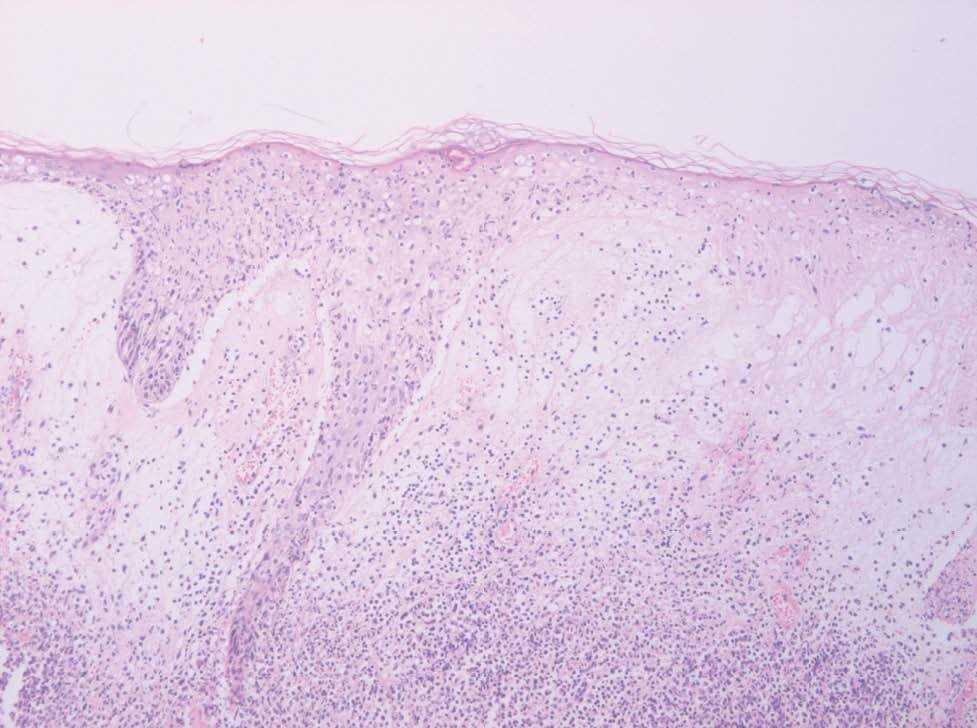
Fig 3.--Biopsia de la lesión.
DIAGNOSTICO
Pioderma gangrenoso (PG) asociado a síndrome mielodisplásico.
HISTOPATOLOGIA
La biopsia de la lesión mostraba un intenso infiltrado en dermis compuesto fundamentalmente por neutrófilos con formación de abcesos, junto con prominente edema en la unión dermoepidérmica con despegamiento de la misma, así como necrosis epidérmica en el borde de la lesión.
EVOLUCION Y TRATAMIENTO
Con el diagnóstico de PG, basándose en los criterios clínicos e histopatológicos se inició tratamiento con deflazacort por vía oral a dosis de 30 mg/día en pauta descendente. En ese momento había aparecido una nueva lesión en el dorso de la mano izquierda clínicamente compatible con PG. Al mes de tratamiento todas las lesiones se encontraban en fase residual. Durante los controles posteriores apareció una nueva lesión, de las mismas características, en el borde lateral de la mano izquierda, que nuevamente respondió al tratamiento con deflazacort (misma pauta).
COMENTARIO
El PG es una dermatosis neutrofílica, idiopática e infrecuente, caracterizada por la formación de una o múltiples ulceraciones cutáneas de bordes violáceos, socavados y rodeados por un halo inflamatorio 1-3.
El diagnóstico se realiza basándose en la clínica, junto con una histopatología compatible y la exclusión de otras causas de úlceras 2.
En un 50-70 % de los casos se encuentra asociado a procesos sistémicos de muy diversa índole. Entre los procesos hematológicos se incluyen la leucemia mieloide y linfoide aguda, síndromes mieloproliferativos, gammapatía monoclonal (sobre todo de clase IgA), mieloma y macroglobulinemia de Waldenstrom. De forma más infrecuente se encuentran los linfomas de Hodgkin, no hodgkinianos y los linfomas cutáneos de células T. Dentro de la enfermedad inflamatoria intestinal (EII) se encuentra asociación con colitis ulcerosa, enfermedad de Crohn, diverticulitis y enteritis regional. Entre las enfermedades reumatológicas destaca la asociación con la artritis reumatoide (AR), la poliartritis seronegativa de pequeñas articulaciones y otras espondiloartropatías seronegativas incluyendo la artritis psoriásica. El PG también se ha visto en el seno de hepatitis crónica activa, cirrosis biliar primaria y colangitis esclerosante. Una variante vesiculopustular ha sido observada en pacientes que presentan de forma simultánea colitis ulcerosa y enfermedades hepáticas. Se ha descrito su asociación con diversas alteraciones de la función inmune como la infección por virus de la inmunodeficiencia humana (VIH), HTLV-III, hipogammaglobulinemia congénita y adquirida, síndrome de hiperIgE, así como diversas inmunodeficiencias celulares y debidas a fármacos. Algunos tumores sólidos (colon, vejiga, pulmón, próstata, bronquios, ovarios y suprarrenal) también se han visto en relación con el PG, así como la toma de fármacos como interferón alfa, sulpiride y factor estimulador de colonias. Otras asociaciones menos frecuentes son enfermedades tiroideas, enfermedad pulmonar obstructiva crónica, hidrosadenitis supurativa, acné conglobata, sarcoidosis, gastritis atrófica, diabetes mellitus, lupus eritematoso, arteritis de Takayasu, dermatomiositis, enfermedad de Wegener, sordera neurosensorial y hemoglobinuria paroxística nocturna. Entre todos ellos, la EII y la artritis son los más comunes 3,4. Por otro lado, en los síndromes mielodisplásicos encontramos asociación con otras dermatosis neutrofílicas, como el síndrome de Sweet o el eritema elevatum diutinum, pudiendo aparecer de forma simultánea o por separado 5.
Se describen 4 variantes clínicas de PG. La forma clásica es el PG ulcerativo, que se presenta como grandes úlceras de bordes violáceos socavados, de forma preferente en extremidades inferiores y tronco. Se ha encontrado una mayor asociación de esta forma con la enfermedad inflamatoria intestinal.
El PG ampolloso aparece de forma preferente en áreas extensoras de brazos y cara, puede cursar con fiebre y artralgias, parece que puede haber un cierto solapamiento con el síndrome de Sweet y se asocia frecuentemente con enfermedades hematológicas malignas, curando típicamente al resolverse el proceso hematológico. Cuando se asocia a leucemia a menudo indica un peor pronóstico.
El PG pustuloso aparece como pústulas de pequeño tamaño, dolorosas, en tronco y extremidades inferiores, en ocasiones con distribución simétrica. Se asocia a EII, así como a infecciones persistentes de la mucosa respiratoria (fibrosis quística).
El PG vegetante se localiza generalmente en el tronco, como una úlcera crónica no dolorosa, sin borde violáceo y de lenta evolución. Generalmente no se encuentra enfermedad sistémica asociada. A menudo responde a terapias menos agresivas, tópicas e intralesionales 3,6.
Los corticoides orales (prednisona 1-2 mg/kg/día) o en bolos (metilprednisona 10-20 mg/kg) han sido durante muchos años el tratamiento de primera línea, siendo éstos especialmente útiles cuando el PG se asocia a enfermedades sistémicas como la EII o AR. La ciclosporina a dosis de 6-10 mg/kg/día, asociada o no a corticoides, ha sido propuesta como tratamiento de primera elección por varios autores. En los casos de PG limitado o localizado se puede considerar la realización de tratamientos tópicos con cromoglicato sódico, triamcinolona intralesional, mesalazina o tacrolimus 0,1 %. En los casos de enfermedad extensa, con varias lesiones, o falta de respuesta a los tratamientos previamente citados está indicada la realización de terapias combinadas con agentes inmunosupresores (azatriopina, ciclofosfamida, clorambucil o mofetil micofenolato), agentes inmunomoduladores (talidomida, infliximab, interferón alfa o inmunoglobulinas por vía intravenosa), o agentes antimicrobianos/antiinflamatorios (dapsona, clofacimina, colchicina o minociclina) 6-9.
Declaración de conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
C. Bahillo Monné. Servicio de Dermatología. Hospital Virgen
de la Salud. Avda. Barber 30. 45004. Toledo. España.
consbahillo2003@yahoo.es
Recibido 12 de julio de 2005.
Aceptado 24 de marzo de 2006.


