


Autoinflammatory type I interferonopathies are disorders characterized by the presence of a type I interferon (IFN) signature in peripheral blood and varying degrees of systemic inflammation. Within this rare group of diseases, stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI), chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature/proteasome-associated autoinflammatory syndrome (candle/praas), and aicardi-goutières syndrome (ags) are the most prevalent.1,2
In this article, we present a typical case of SAVI syndrome with a genetic variant not previously described in the literature.
A 75-year-old woman described a past medical history of painful skin lesions on acral regions since early childhood. Specifically, during the cold months of the year, erosions and ulcers appeared on the dorsal and palmar regions of the distal fingers and toes. As an accompanying symptom, she reported dyspnea with significant exertion since childhood, preventing her from engaging in physically demanding activities throughout her life. She had consulted multiple specialists, with the final diagnosis of idiopathic perniosis. In the differential diagnosis, other causes of perniosis were excluded, including systemic lupus erythematosus, cryoglobulinemia, hematologic disorders (including monoclonal gammopathies), trauma, and medication-induced etiologies.
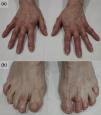
Upon examination (Fig. 1), erythematous and edematous painful plaques were observed on superficial and deep palpation, with some superficial erosions on the fingers (Fig. 1a) and toes (Fig. 1b). A skin biopsy of the perniosiform lesions could not be performed following the patient's denial to undergo the procedure. No other mucocutaneous lesions were observed.

Clinical appearance of the lesions. In addition to the characteristic lesions of osteoarthritis (Heberden's and Bouchard's nodes), erythema and edema were observed in the distal regions of the hands [panel (a)] and feet [panel (b)]. Lesions were most pronounced on the 2nd and 4th toes of the left foot and the 5th toe of the right foot, corresponding to sites of severe ulceration in previous years.
During the directed medical history, she reported that 2 of her 3 children had similar symptoms and that, according to available information, her deceased mother had also experienced this condition at a young age. Suspecting genetically originated perniosis, a genetic study was conducted to rule out other causes, including SAVI syndrome, familial lupus pernio, cryopyrinopathies, or COPA syndrome. The genetic analysis reported the c.497G>A, p.Gly166Glu (NM_198282.4) variant in exon 5 of the STING1 gene, thus confirming the diagnosis of SAVI syndrome. Subsequently, the study confirmed positive results in her 2 symptomatic children and negative results in the asymptomatic child. Additionally, a high-resolution chest computed tomography (HRCT) scan yielded normal results. Finally, pulmonary function tests (PFTs) reflected a mild restrictive pattern. Given the relatively non-disabling nature of the symptoms and after reaching a consensus with the patient regarding therapeutic options, clinical monitoring with an emphasis on avoiding cold exposure was selected.
Stimulator of interferon genes (STING), encoded by the transmembrane protein 173 gene (TMEM137), plays a crucial role in activating the IFN response. Pathogenic heterozygous gain-of-function variants in STING1 lead to constitutive activation, resulting in the clinical syndrome known as SAVI, first described in 2014.3 Although it follows an autosomal dominant pattern, de novo mutations have been reported.4 Its clinical signs include acral perniosis-like skin lesions, nasal septum, and auricular cartilage necrosis. Associated symptoms may include intermittent low-grade fever, recurrent cough and dyspnea indicating interstitial lung disease, and occasional growth failure and joint involvement.3,4 Diagnosis is suspected in refractory perniosis-like clinical presentation, especially when associated with interstitial lung disease and a family history of autosomal dominant inheritance. Genetic testing confirms the diagnosis, with around 20 reported mutations, with p.V155M being the most common.4 However, the patient exhibited a mutation not yet described in international registered databases, specifically variant in heterozygosity c.497G>A, p.Gly166Glu (NM_198282.4) in exon 5 of the STING1 gene. Differentiation from other causes of perniosis or genetic perniosis syndromes, such as COPA syndrome, familial lupus pernio, or cryopyrinopathies is essential.2,4 Prognosis is usually influenced by pulmonary involvement, thus, pulmonary assessment with PFTs and HRCT for all patients is recommended. Treatment involves cold avoidance, and systemic corticosteroids may provide partial responses.4,5 Recently, JAK inhibitors have proven safe and effectivue in some cases.5 In conclusion, we present a case of a typical SAVI syndrome with a novel mutation. Awareness of this condition is crucial in refractory perniosis, necrosis of nasal or auricular cartilage, dyspnea, and joint disease since childhood with autosomal dominant inheritance.
Conflict of interestThe authors declare that they have no conflict of interest.


