




















INTRODUCCION
Lichtenstein introdujo en 1953 el término de «histiocitosis X» para agrupar tres entidades con características clínicas e histopatológicas semejantes que habían sido descritas con anterioridad: el granuloma eosinófilo, la enfermedad de Letterer-Siwe y la enfermedad de Hand-Schüller-Christian 1. Posteriormente se abandonó este término al ser identificadas las células implicadas, y se acuñó el nombre de histiocitosis de células de Langerhans (HCL) 2.
La característica común de estas entidades es la proliferación de células de Langerhans y su infiltración en diferentes tejidos. En el momento actual, bajo este término se engloban cuatro formas de presentación clínica principales: la enfermedad de Letterer-Siwe, la enfermedad de Hand-Schüller-Christian, el granuloma eosinófilo y la enfermedad de Hashimoto-Pritzker. La HCL se corresponde con el tipo I de la clasificación de las histiocitosis del Writting Group of the Histiocyte Society publicada en 1987 3 (tabla 1). Este mismo grupo ha publicado posteriormente otra clasificación en la que incluyen la HCL dentro de las histiocitosis de comportamiento biológico variable 4 (tabla 2).


CÉLULAS DE LANGERHANS
Las células de Langerhans pertenecen a la familia de las células dendríticas, un grupo de células presentadoras de antígeno que se originan en la médula ósea a partir de un antecesor CD34+, común con el sistema fagocítico mononuclear 4. Las células dendríticas comparten propiedades con monocitos y macrófagos, pero poseen distintiva citomorfología, inmunohistoquímica, ultraestructura e inmunofenotipo, lo que permite su identificación 5. Las células de Langerhans se encuentran localizadas habitualmente en la epidermis, los ganglios linfáticos, el bazo, el timo, las mucosas del epitelio intestinal y respiratorio, y el cérvix 6. En la epidermis se sitúan sobre la capa basal, lo cual las diferencia habitualmente de los melanocitos.
Poseen numerosos marcadores de superficie, en relación a su función de células presentadoras de antígeno, incluyendo el antígeno de histocompatibilidad (HLA) de clase I y II, receptores para la región Fc de la inmunoglobulina G, un receptor de alta afinidad para la inmunoglobulina E (FcεR1) y receptores para el complemento (C3bi/CR III) 7. El receptor CD1a las distingue de otras células dendríticas procesadoras de antígeno y del resto de los histiocitos. Sintetizan interleucinas (IL-1, IL-6) y factor de necrosis tumoral a (TNF-α), entre otras citocinas.
Ultraestructuralmente poseen en el citoplasma unas organelas características con forma de raqueta de tenis llamadas gránulos de Birbeck. Son invaginaciones de la membrana plasmática, implicadas en el procesamiento antigénico. El Lag es un antígeno presente en la membrana de los gránulos de Birbeck, y su identificación puede utilizarse para confirmar la presencia de células de Langerhans en los tejidos 8,9.
Como hemos apuntado anteriormente, son las principales células presentadoras de antígeno presentes en la epidermis (constituyendo un 2 % del total celular) y son esenciales para la integridad del sistema inmunitario cutáneo 9. Después de un contacto con un antígeno, estas células migran a los ganglios linfáticos regionales, donde presentan el antígeno a los linfocitos T CD4+ en la región paracortical. Desempeñan un papel importante en la patogenia de la dermatitis alérgica de contacto y un papel menor en otras dermatosis inflamatorias como el liquen ruber (rojo) plano. Constituyen las células diana en el rechazo a los injertos, y en la enfermedad injerto contra huésped 10. Con frecuencia aparecen aumentadas en número en la epidermis que recubre numerosos tumores 9. Se destruyen por la irradiación o por la luz ultravioleta 7.
En la HCL, las células de Langerhans mantienen el perfil antigénico de superficie de las células dendríticas normales. Según algunas publicaciones también mantienen su función como células presentadoras de antígeno 6, mientras que para otras esta función se encuentra atenuada 8,11.
EPIDEMIOLOGIA
La incidencia de la HCL es desconocida, sobre todo en relación a la gran heterogeneidad clínica. En la población general se estima una incidencia en torno a 2-5 casos por millón de habitantes al año 6,12. Es más frecuente en el sexo masculino, sobre todo en la infancia, puesto que en los adultos las series son muy contradictorias 13,14. Suele manifestarse en niños, pero actualmente se han descrito series largas en adultos 13-16.
Se han identificado como antecedentes epidemiológicos de interés la exposición de los padres a diversos solventes, la historia familiar de tumores benignos, infecciones urinarias durante el embarazo, transfusiones sanguíneas posnatales, infecciones perinatales e historia familiar positiva de enfermedades tiroideas, desconociéndose en el momento actual el significado de estos hallazgos 8,15,17-19.
ETIOPATOGENIA
La etiopatogenia de esta enigmática entidad no está bien aclarada. En la actualidad, la mayor parte de los investigadores cree que se produce una alteración en la regulación del sistema inmunológico en estos pacientes 8,12,20-23. La presencia de agregados de otras células inmunológicamente activas en las lesiones, las anomalías encontradas en el timo, la disminución en el número de los linfocitos T supresores CD8+ y el aumento en la síntesis de varias citocinas (factor estimulador de macrófagos y granulocitos, IL-1, IL-3, IL-8, IL-10, TNF-α, interferón-γ) sugieren una respuesta fisiológica exagerada de las células de Langerhans a un antígeno no identificado o, por el contrario, una apropiada respuesta a señales anormales procedentes de otras células del sistema inmunológico 21. El factor estimulante de colonias de granulocitos y macrófagos parece tener una función preponderante en el reclutamiento, la activación, la proliferación y la supervivencia de las células de Langerhans, por lo que puede constituir un marcador biológico de la masa tumoral 24. Aunque actualmente se cuenta con numerosos datos sobre los mediadores celulares involucrados, no se ha podido demostrar la existencia de ningún defecto primario en el sistema inmunológico responsable del desencadenamiento de la HCL 25.
Se ha comprobado con técnicas de citometría de flujo la monoclonalidad en las células de Langerhans del infiltrado, sugiriéndose una etiología neoplásica 25-27. Sin embargo, existen proliferaciones clonales que son benignas. Se puede añadir que las figuras mitóticas son muy escasas 5,20,28 y que se ha descrito una resolución espontánea de las lesiones, incluso en afectaciones multisistémicas, lo cual apoyaría un proceso reactivo más que neoplásico 29. Estos datos requieren que la existencia de monoclonalidad de las células deba ser cuidadosamente considerada. Algunos autores sugieren que la HCL pueda tratarse de un proceso neoplásico con una gran variabilidad en cuanto a su comportamiento biológico, y a su intensidad clínica 4,25.
Leahy et al 30 detectaron mediante la técnica de la reacción en cadena de la polimerasa la presencia del virus herpes tipo 6 en las lesiones de la HCL. Posteriormente otros autores no han conseguido demostrar estos hallazgos 31. No se ha detectado ningún otro genoma viral en las lesiones y su implicación en el desencadenamiento de esta entidad sigue en estudio 25.
MANIFESTACIONES CLINICAS
La HCL puede afectar a uno o varios órganos y a una o varias localizaciones, lo cual es muy importante para el pronóstico y el tratamiento. Los órganos afectados con más frecuencia son los huesos, la piel, los ganglios linfáticos, la médula ósea, el hígado, el bazo y el SNC. La enfermedad de Letterer-Siwe y la de Hand-Schüller-Christian son casi exclusivas de la infancia, aunque se ha descrito su presentación en pacientes adultos 32. En estos pacientes es más frecuente la afectación aislada de un órgano que la forma multiorgánica. La incidencia en ancianos es excepcional y en ellos el diagnóstico de sospecha clínica es muy difícil, aunque suele asociarse a un mejor pronóstico 33,34.
Enfermedad de Letterer-Siwe
La enfermedad de Letterer-Siwe representa la forma aguda y multisistémica de la HCL. En la mayoría de los casos se manifiesta antes de los 2 años de edad, con un cuadro que recuerda a una infección sistémica, o a un tumor maligno y suele acompañarse de fiebre, adenopatías y hepatoesplenomegalia. Se han publicado casos congénitos y familiares 12.
Las manifestaciones cutáneas son muy frecuentes y pueden ser el primer signo de la enfermedad en aproximadamente el 50 % de los casos 12. Aparecen pápulas rosado-amarillentas descamativas, con tendencia a la formación de costras en su superficie, localizadas característicamente en áreas seborreicas en el tronco, cara y cuero cabelludo (figs. 1, 2 y 3). Otra manifestación típica la constituyen las erosiones crónicas en axilas, ingles, retroauriculares y región anogenital (figs. 4 y 5), y con frecuencia es la forma de inicio en adultos (figs. 6 y 7) 33. La distribución «en camiseta» de las lesiones se ha descrito como patognomónica (fig. 8) 12. Cuando predomina la púrpura secundaria a la trombocitopenia se considera un signo pronóstico muy desfavorable 35. Otra forma frecuente de presentación consiste en la aparición de uno o varios nódulos, con tendencia a la ulceración, y excepcionalmente a la formación de grandes masas tumorales 36. En niños muy pequeños puede desarrollarse una erupción vesículo-pustulosa que obliga a realizar el diagnóstico diferencial con una miliaria, un eczema, una escabiosis o, incluso, una varicela. La participación de palmas y plantas se confunde con lesiones eczematosas o con infecciones por dermatofitos. La afectación ungueal está infradiagnosticada y suele atribuirse a morderse las uñas. Aparece en forma de paroniquia, destrucción de pliegues ungueales, onicólisis, hiperqueratosis subungueal o estrías purpúricas. Algunos autores han sugerido el valor pronóstico desfavorable de la afectación ungueal, pero el reducido número de casos descritos no permite llegar a ninguna conclusión 37,38. En niños de raza negra, las lesiones aparecen hipopigmentadas 7. No es frecuente la implicación de las mucosas (fig. 9) 12.
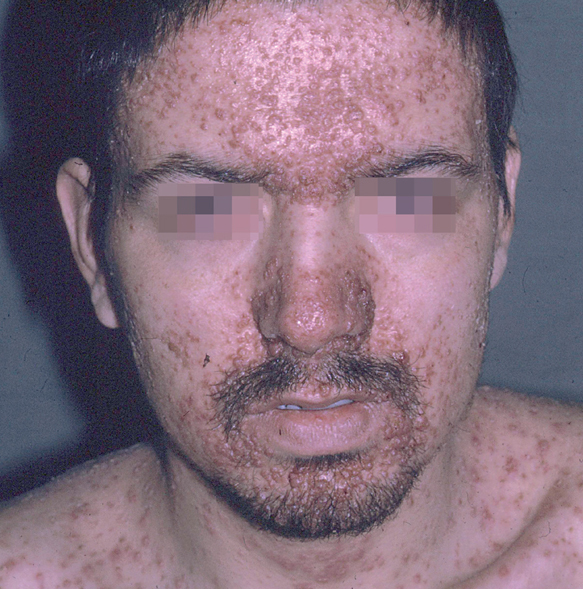
Fig. 1.--Pápulas eritematosas confluentes en la cara, con predominio en las zonas seborreicas en la enfermedad de Letterer-Siwe.
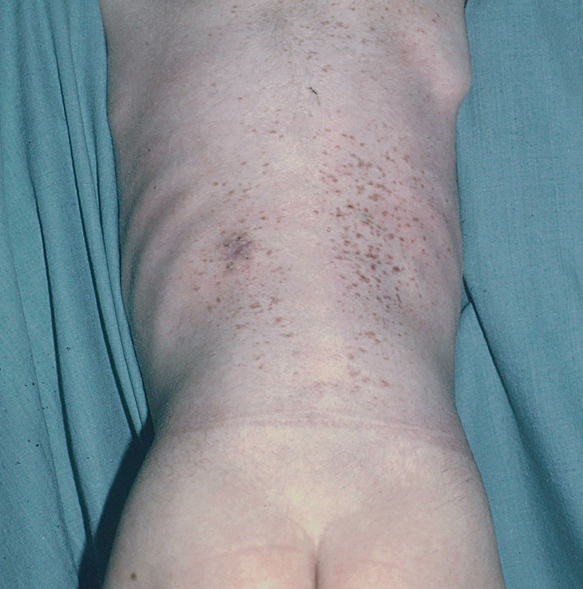
Fig. 2.--Pápulas eritematosas localizadas en la espalda de una niña de 2 años en la enfermedad de Letterer-Siwe.
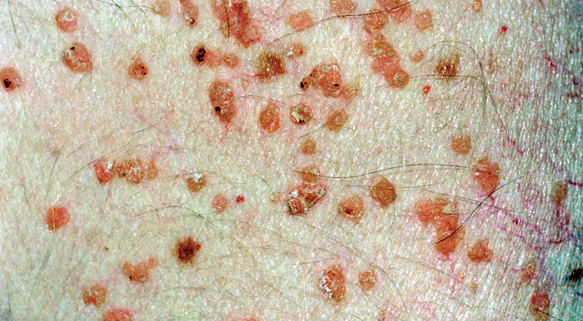
Fig. 3.--Detalle de las lesiones características de la HCL, con pápulas eritematosas con costras en la superficie.
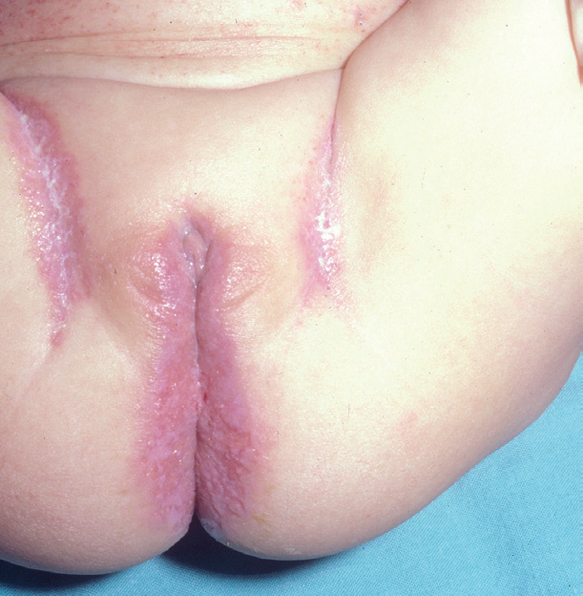
Fig. 4.--HCL. Lesiones en ingles.
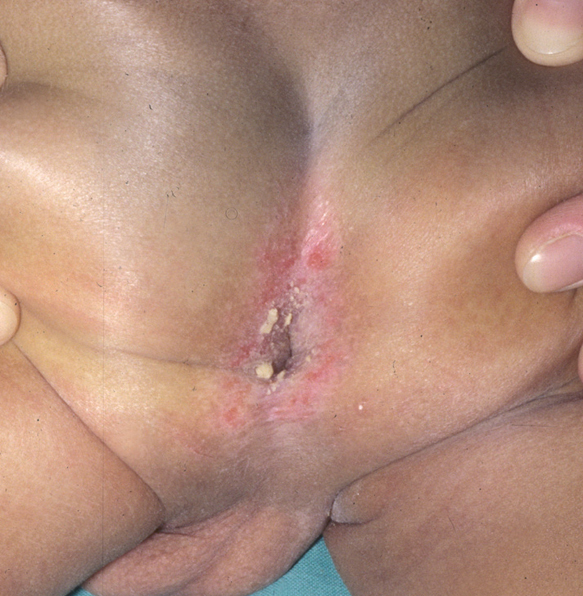
Fig. 5.--HCL. Lesiones anogenitales.
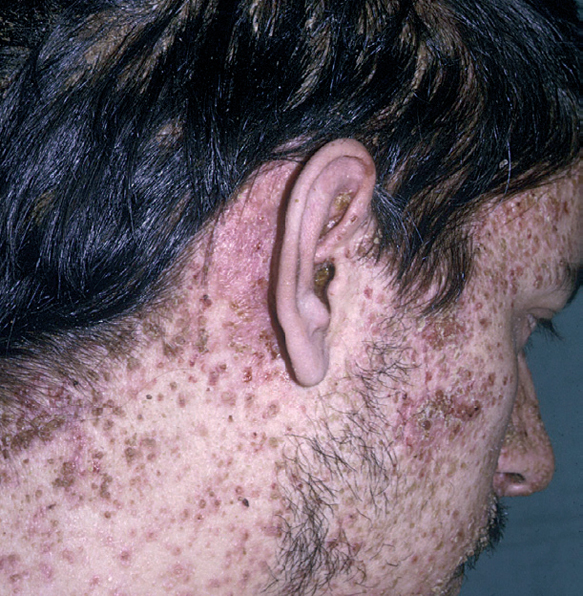
Fig. 6.--En la enfermedad de Letterer-Siwe. Erosiones retroauriculares y pápulas eritematosas costrosas en el cuero cabelludo y en las caras laterales del cuello.
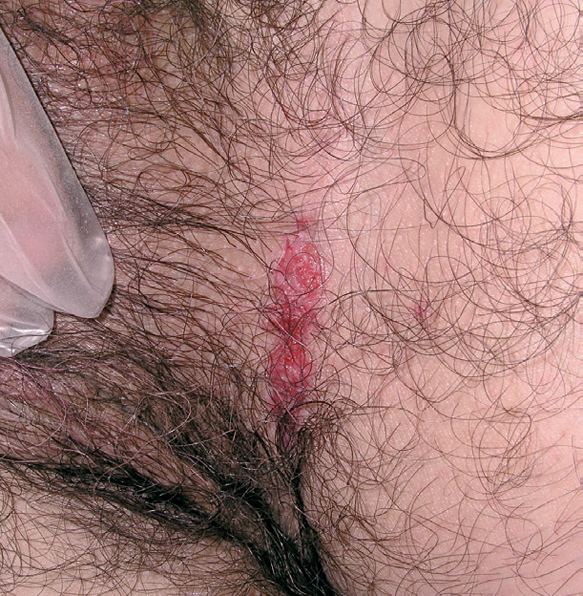
Fig. 7.--Lesiones erosivas inguinales, características en las formas de inicio en adultos.
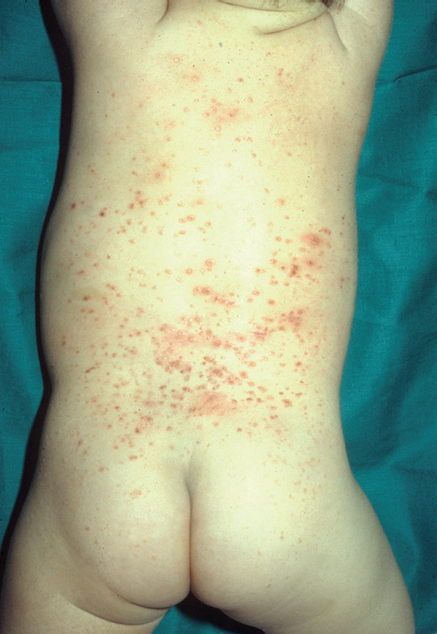
Fig. 8.--Lesiones en el tronco.
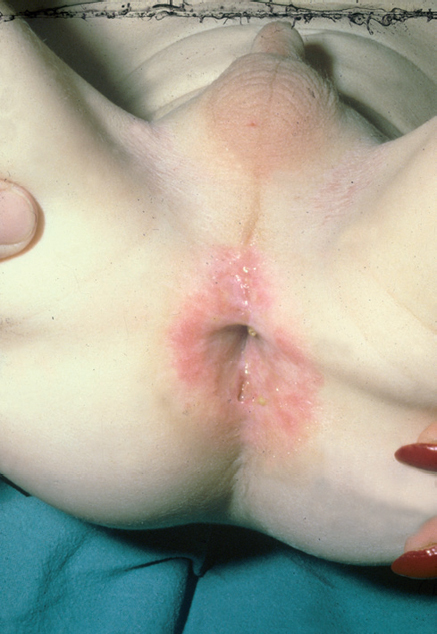
Fig. 9.--Lesiones mucosas en HCL.
Dentro de las afectaciones extracutáneas destaca la ósea, en más del 60 % de los casos 12,15. Las localizaciones habituales son el cráneo, las costillas y las vértebras y, en menor proporción, el húmero y el fémur 13. Son lesiones osteolíticas asintomáticas o dolorosas localmente, que pueden acompañarse de una inflamación del tejido cutáneo suprayacente. Generalmente son múltiples, y aparecen gradualmente. Si la localización es periorbitaria, puede aparecer proptosis, en el hueso maxilar, pérdidas dentarias y en la apófisis mastoides, otitis media. De manera excepcional se detecta hipercalcemia secundaria a lesiones óseas líticas difusas 39.
La afectación pulmonar ocurre con mayor frecuencia en los adultos, puesto que el tabaco parece ser un importante cofactor etiológico 8,15. Se describe como un patrón quístico o nodular, que evoluciona a fibrosis pulmonar intersticial y se manifiesta clínicamente en forma de neumotórax espontáneo, disnea o taquipnea. Su diagnóstico se establece mediante un lavado broncoalveolar, con más del 1 % de células CD1a+.
Se pueden encontrar linfadenopatías, sobre todo en localización cervical, generalmente asociadas a afectación multiorgánica 8. El hígado y el bazo son dos órganos de «riesgo», y su afectación se considera un signo de pronóstico desfavorable, particularmente cuando aparece ictericia, u otros signos de insuficiencia hepática. Aparecen involucrados en estadios tardíos en pacientes con enfermedad multisistémica 15. La participación de la médula ósea ocurre en pacientes con una infiltración difusa de otros órganos. La leucocitopenia establece un mal pronóstico 12.
El diagnóstico de la afectación intestinal es difícil porque acontece de forma intermitente, con una sintomatología de diarrea o malabsorción, y requiere una confirmación endoscópica. La afectación de la mucosa oral debe hacer investigar de forma sistemática una eventual afectación digestiva, más frecuente en estos casos 8,24,40. Por último, en el SNC la manifestación más frecuente y característica es la diabetes insípida.
Enfermedad de Hand-Schüller-Christian
La enfermedad de Hand-Schüller-Christian representa la forma crónica y multifocal de la HCL. Se manifiesta entre los 2 y los 6 años de edad en el 70 % de los casos 22. Se caracteriza por la clásica tríada de lesiones óseas, exoftalmos y diabetes insípida. Es infrecuente encontrar las tres en el mismo paciente, lo cual sólo ocurre en el 10 % de los casos 9. Las lesiones óseas son las manifestaciones más frecuentes. Aparecen en un 80 % de los pacientes, sobre todo en los huesos craneales, especialmente en la región temporoparietal, con lesiones osteolíticas que adoptan una apariencia «en mapa». La presencia de diabetes insípida se considera un signo pronóstico favorable 22. El exoftalmos ocurre por lo general en fases avanzadas de la enfermedad y afecta al 10-30 % de los pacientes.
La afectación cutánea se produce en un tercio de los casos. Su aspecto clínico es similar al descrito en los pacientes con la enfermedad de Letterer-Siwe. Pueden observarse lesiones papulonodulares frecuentemente ulceradas en la mucosa oral o en la región vulvar (fig. 10) 12.
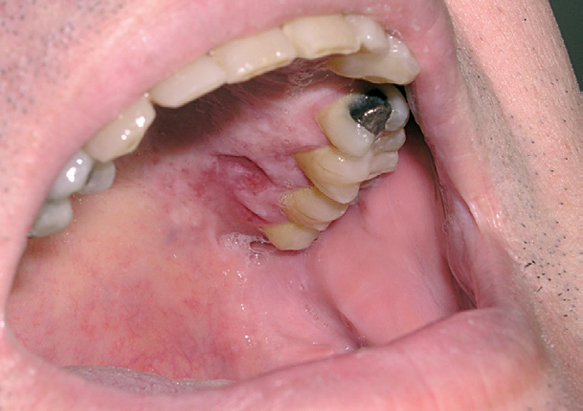
Fig. 10.--Lesiones ulceradas en la mucosa oral en un paciente con enfermedad de Hand-Schüller-Christian.
Granuloma eosinófilo
Constituye la forma localizada y benigna de la HCL. Generalmente se manifiesta entre los 5 y los 30 años de edad y afecta con mayor frecuencia a los varones 22. La afectación ósea es la más llamativa, a menudo en forma de lesiones solitarias, con el siguiente orden de frecuencia: cráneo, costillas, columna vertebral, pelvis, escápula y huesos largos 12. Las lesiones cutáneas son muy infrecuentes. Cuando aparecen generalmente son nódulos ulcerados bien delimitados, rebeldes al tratamiento, localizados en la mucosa oral o genital, o retroauriculares. Se ha descrito el granuloma eosinofílico periorificial característicamente en series de adultos.
Hashimoto-Pritzker
La variante congénita se denomina enfermedad de Hashimoto-Pritzker, o reticulohistiocitosis congénita autoinvolutiva 41. Las lesiones cutáneas están presentes desde el nacimiento o en los primeros meses de vida, en forma de nódulos rojo-parduscos múltiples o solitarios, con una tendencia variable a la ulceración y a la formación de costras 42,43. Suelen mejorar espontáneamente durante el primer año de vida, localizándose en cualquier parte de la superficie corporal, pero sin afectación orgánica. El compromiso de las mucosas es excepcional. Pueden observarse lesiones urticariformes con un signo de Darier positivo, debido a la presencia de mastocitos en el infiltrado acompañante 44. En casos aislados, se ha descrito una afectación sistémica, que también se resuelve espontáneamente. Es obligado, sin embargo, realizar un seguimiento de estos niños durante varios años, puesto que se han publicado casos tardíos de recaídas o de aparición de manifestaciones extracutáneas 7. Los hallazgos que se encuentran en la microscopia óptica y en las tinciones inmunohistoquímicas pueden ser idénticos a los de la HCL. En los estudios con el microscopio electrónico se observan gránulos de Birbeck y/o cuerpos densos mieliniformes en menos del 40 % de las células histiocitarias, mientras que en la HCL los encontramos en más del 50 % de los casos 42,45.
En ocasiones, las manifestaciones clínicas son atípicas, y no cumplen los criterios de ninguna de estas cuatro entidades. Por esta razón, existe una creciente tendencia a englobarlas bajo el término de HCL, y clasificarlas en función de su extensión y de la rapidez de su instauración (tabla 3) 7,46. Se han descrito presentaciones cutáneas poco frecuentes, que incluyen lesiones semejando pioderma gangrenoso, granuloma anular, amebiasis, xantomas eruptivos, picaduras, angiomas rubí, verrugas planas y enfermedad de Darier 47-53. También se ha descrito una variante con lesiones tipo cutis laxa, en una paciente obesa con histiocitosis X y afectación cutánea pura 54.

La asociación de la HCL con neoplasias malignas ha sido apuntada en varias series de pacientes, siendo mayor de la esperado por el azar. Estas neoplasias incluyen linfomas, leucemias, carcinoma pulmonar, carcinoma tiroideo, carcinoma de mama, astrocitoma, glioblastoma, meduloblastoma, meningioma, ependimoma, condrosarcoma, retinoblastoma, carcinoma basocelular, carcinoma lingual, apudoma, carcinoma hepatocelular, carcinoma gástrico, sarcoma de Ewing y osteosarcoma 5,55-57. Egeler et al 5,58 han descrito dos grupos diferentes de asociación en estos pacientes. Por una parte, la íntima y simultánea aparición con los linfomas y con el carcinoma pulmonar sugiere que estén relacionados de forma reactiva. Sin embargo, en los pacientes con leucemia y otros tumores sólidos, el periodo de latencia de la neoplasia maligna después del diagnóstico de la HCL sugiere fuertemente que la terapia empleada con quimioterapia y radioterapia sea la responsable de incrementar el riesgo de padecer estos tumores. Otros autores han descrito la asociación de la HCL con leucemias de la línea mielomonocítica de presentación muy cercana en el tiempo y sin tratamiento quimioterápico previo y sugieren un origen común para ambos procesos por alteraciones de la célula madre pluripotencial CD34+*33.
DIAGNOSTICO
El diagnóstico se plantea a partir de las manifestaciones clínicas y para confirmarlo es necesario biopsiar las lesiones sospechosas y realizar tinciones específicas para CD1a, y S100, o demostrar la presencia de gránulos de Birbeck en el citoplasma con el microscopio electrónico, o con marcadores específicos 4,6,8.
La evaluación completa de cualquier paciente, ya se trate de una afectación única, o de múltiples órganos, debería incluir un hemograma completo, determinación del tiempo de protrombina y del tiempo de tromboplastina parcial, pruebas de función hepática, proteinograma, recuentos de células T y B, análisis de subgrupos de células T, análisis de orina, serie ósea y radiografía de tórax. Si existe una afectación más específica es necesario realizar tomografías en diversas localizaciones, aspirados de médula ósea, y/o prueba de restricción hídrica, si se sospecha una diabetes insípida (tabla 4) 8,22.

HISTOPATOLOGIA
Las células de Langerhans son largas células ovoidales, de unos 12-15 mm de diámetro, de bordes mal definidos, con abundante citoplasma eosinófilo y un núcleo excéntrico en forma de «grano de café» (fig. 11). Como hemos adelantado, presentan marcadores inmunohistoquímicos característicos como S100, CD1a, CD45, HLA-DR, CD4, CD2 y T200 (fig. 12) 12. Comparten con el sistema monocito/macrofágico algunos antígenos comunes: CD1a, CD11 y CD45.
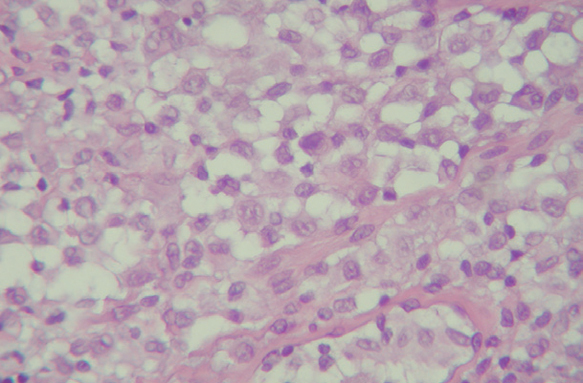
Fig. 11.--HCL cutánea, mostrando las células de Langerhans con su característico citoplasma eosinófilo y núcleo en forma de «grano de café». (Hematoxilina-eosina, x40.)
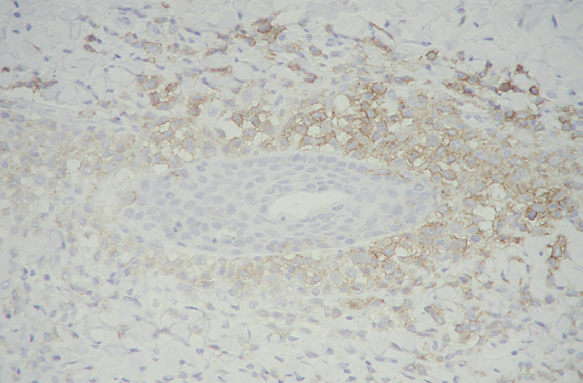
Fig. 12.--Inmunohistoquímica con CD1a, que tiñe selectivamente las células de Langerhans. (CD1a, x40.)
Existen algunos marcadores que diferencian a las células que se acumulan en esta enfermedad de las células de Langerhans de la piel normal que son la fosfatasa alcalina placentaria, la aglutinina del cacahuete, el receptor para el interferón-g y los marcadores CD11c, CDW32, CD68, CD14 22,31,59. Son negativas para Mac-387, que tiñe las células del sistema fagocítico mononuclear. Tampoco expresan CD34 ni MS-1, que son marcadores de células endoteliales sinusoidales y de macrófagos dendríticos perivasculares.
En la HCL el infiltrado se localiza normalmente en la dermis adventicial, y puede invadir la epidermis, produciendo clínicamente lesiones erosivas (figs. 13 y 14). En las formas nodulares puede invadir la hipodermis. En pacientes adultos se ha descrito la distribución del infiltrado alrededor de los anejos (figs. 15 y 16) 13. Aparecen otras células inflamatorias en número variable, sobre todo neutrófilos, eosinófilos, linfocitos y mastocitos y, en menor medida, células gigantes multinucleadas. De forma clásica se han descrito tres subtipos principales: proliferativo, granulomatoso y xantomatoso, que se intentan correlacionar con las diferentes formas clínicas. Histológicamente no existen diferencias entre la HCL y la reticulohistiocitosis congénita autoinvolutiva 7.
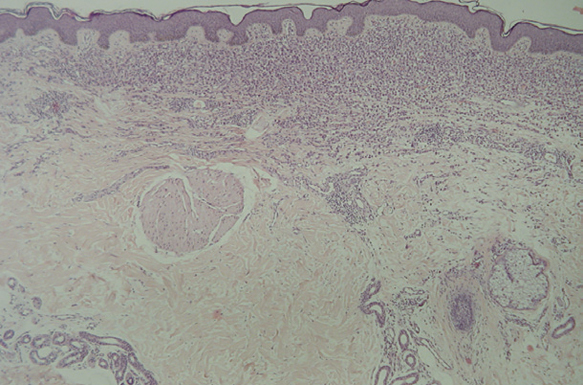
Fig. 13.--Infiltrado en placa subepidérmico conformado por células de Langerhans. (Hematoxilina-eosina,x10.)
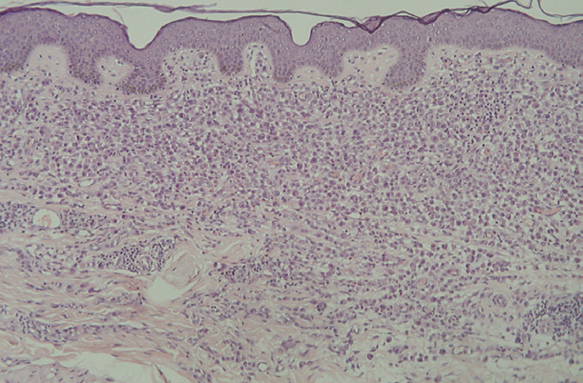
Fig. 14.--A mayor aumento, detalle del infiltrado en dermis reticular. (Hematoxilina-eosina, x20.)
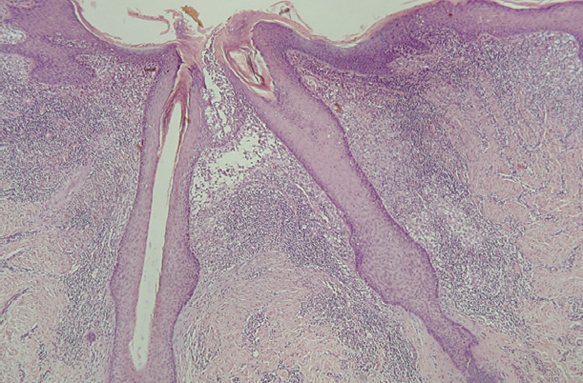
Fig. 15.--Infiltrado de células de Langerhans rodeando los folículos pilosos. (Hematoxilina-eosina, x10.)
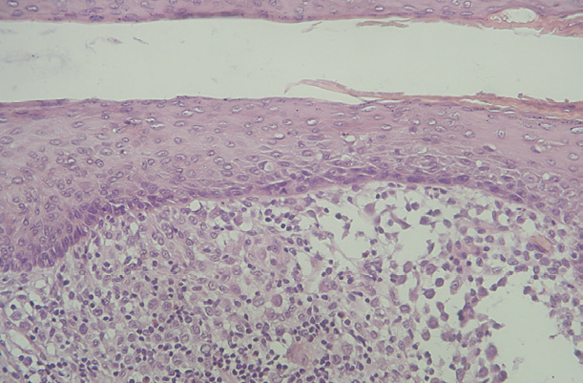
Fig. 16.--Detalle del infiltrado rodeando los anejos. (Hematoxilina-eosina, x20.)
Con el microscopio electrónico se pueden observar los gránulos de Birbeck, que no están presentes en todas las células en la HCL. Son numerosos en las lesiones cutáneas, pero escasos en las lesiones viscerales 12.
Aunque los cambios histopatológicos no constituyen un factor pronóstico, la fuerte expresión del antígeno nuclear proliferante en las células de Langerhans parece conferir una peor evolución 13.
DIAGNOSTICO DIFERENCIAL
En el diagnóstico diferencial se incluye exclusivamente el de las lesiones cutáneas de la HCL. En las formas diseminadas conviene realizar el diagnóstico diferencial con la dermatitis seborreica, predominando las lesiones purpúricas en la HCL. Es muy importante pensar en este cuadro ante una dermatitis seborreica rebelde a todo tipo de tratamientos y confirmar el diagnóstico con una biopsia. Deben incluirse en el diagnóstico diferencial los síndromes familiares hemofagocíticos, el síndrome de Rosai-Dorfman y el síndrome hemofagocítico de etiología viral 13. Cuando predominan las lesiones en los pliegues cutáneos hay que excluir otras causas de intertrigo, como la candidiasis. También debe incluirse en los adultos la enfermedad de Darier, con lesiones papulosas más hiperqueratósicas, depresiones puntiformes palmoplantares y una histopatología característica 47. En el liquen nítido las pápulas son brillantes y apenas descamativas y presentan el fenómeno de Koebner 22.
Las lesiones xantomatosas deben diferenciarse de la urticaria pigmentosa, de las histiocitosis sin células de Langerhans y del xantoma diseminado. En las primeras, el signo de Darier es positivo. Por último, en la enfermedad de Hashimoto-Pritzker debemos excluir el granuloma eosinófilo, el xantogranuloma juvenil, la histiocitosis cefálica benigna, la histiocitosis eruptiva generalizada y la urticaria pigmentosa.
PRONOSTICO
La HCL se asocia con un espectro clínico muy amplio y, por lo tanto, con una evolución muy variable. La enfermedad que afecta a un único sistema en general se asocia con buen pronóstico, mientras que en las formas multisistémicas puede ser fatal. Es aconsejable realizar un seguimiento a largo plazo, incluso tras una resolución inicial.
Las causas más frecuentes de muerte son el compromiso pulmonar, medular y las infecciones 15,22. Conviene ser muy cautos a la hora de establecer afirmaciones pronósticas. La mayoría de los niños con afectación cutánea localizada evolucionan bien, pero un pequeño porcentaje puede evolucionar a una afectación multisistémica.
El principal factor pronóstico lo constituye la disfunción de los órganos vitales involucrados, sobre todo el hígado, el pulmón y el sistema hematopoyético 6. También son relevantes:
1.La edad del paciente en el momento del diagnóstico, puesto que los niños menores de 2 años, con enfermedad diseminada, tienen una mortalidad de más del 50 % 7.
2.El número de órganos afectados.
3.La velocidad de extensión de las lesiones.
4.La rapidez de respuesta al tratamiento.
Resulta imposible atribuir una importancia pronóstica a un signo aislado, pero la ictericia, la trombocitopenia, la anemia o la insuficiencia hepática se consideran signos pronósticos desfavorables, mientras que las lesiones de tipo nodular aportan un pronóstico favorable 22.
Son frecuentes las secuelas tardías con incapacidades ortopédicas, deterioro de la audición, alteraciones neurológicas, retraso en el crecimiento, alteraciones pulmonares o cirrosis hepática.
TRATAMIENTO
No existe un claro consenso en cuanto al tratamiento de esta entidad, pese a que se han llevado a cabo estudios para intentar protocolizarlo 60-62. En general, el tratamiento depende de la edad del paciente y del número y disfunción de los órganos afectados.
En las lesiones aisladas, que afectan a la piel o al sistema óseo, está indicada una actitud conservadora. Las lesiones cutáneas con frecuencia no requieren tratamiento, aunque responden bien a los corticoides tópicos o a la mostaza nitrogenada 7,8. La terapia consiste en la aplicación diaria durante 5 días de mostaza nitrogenada diluida en agua hasta una concentración de 20 mg/dl. En los adultos se ha demostrado eficaz el tratamiento con psoraleno y luz ultravioleta (PUVA), y en los casos refractarios se emplean glucocorticoides orales, talidomida o isotretinoína 63-65. En los pacientes con afectación ósea, el tratamiento de elección es el legrado o la escisión quirúrgica, siempre que la localización lo permita. A veces es necesaria la radioterapia. En la infancia pueden utilizarse glucocorticoides intralesionales, para evitar traumatismos óseos.
En los pacientes con enfermedad multisistémica y/o afectación de órganos vitales, un ciclo corto con un solo agente no suele ser suficiente, y se producen recaídas con frecuencia. La mayoría de los estudios emplea en estas circunstancias glucocorticoides asociados a vinblastina por un periodo de entre 6 y 12 meses. Si no responden pueden introducirse regímenes de poliquimioterapia con etopóxido, vincristina, ciclofosfamida, clorambucilo y/o doxorubicina. En casos refractarios existen otras alternativas como ciclosporina, metotrexato o talidomida 8,64,66-68. En la actualidad está en estudio el uso de la 2-clorodesoxiadenosina y de los agentes anti-TNF-α 69-72.
Hay evidencias a favor de que el trasplante de médula ósea o de cordón umbilical pueda ser beneficioso en aquellos casos que no respondan a la quimioterapia 73,74.
AGRADECIMIENTOS
Al los Dres. Lázaro y Lecona, por la ayuda en relación con las ilustraciones de este artículo, y al Dr. Torrelo por su contribución con las fotos clínicas, al tiempo que serviría para expresar mi agradecimiento.


