



La hiperqueratosis filiforme (HF) es una dermatosis infrecuente descrita en la literatura bajo diversos términos1. Su asociación con patología subyacente continúa siendo discutida. Describimos un nuevo caso de HF sin asociación con malignidad subyacente y revisamos los casos existentes en la literatura con lesiones localizadas en palmas y/o plantas y columnas de paraqueratosis. Según la clasificación de las HF establecida por Zarour et al2 y modificada posteriormente por McGovern y Gentry1, estos hallazgos clínicos e histológicos son definitorios de la HF tipo Ia (HF paraqueratósica palmoplantar).
Caso clínico
Un paciente varón de 72 años con antecedentes de espondilolistesis consultó por la presencia de lesiones cutáneas de tres meses de evolución que describía como «espinas» en los dedos de ambas manos, sin otros síntomas acompañantes. Las lesiones le resultaban ligeramente dolorosas a la presión y no se desprendían fácilmente de su base. El paciente no refería alteraciones en la sudoración, antecedentes de exposición a arsénico ni casos similares entre sus familiares.
En la cara palmar de los dedos de ambas manos podían observarse múltiples lesiones filiformes hiperqueratósicas distribuidas irregularmente, de aproximadamente 1-2 mm de altura y coloración amarillenta, algunas de las cuales asentaban sobre tenues máculas eritematoedematosas (fig. 1). En las plantas de ambos pies, cabello y uñas no se detectaron anomalías significativas.
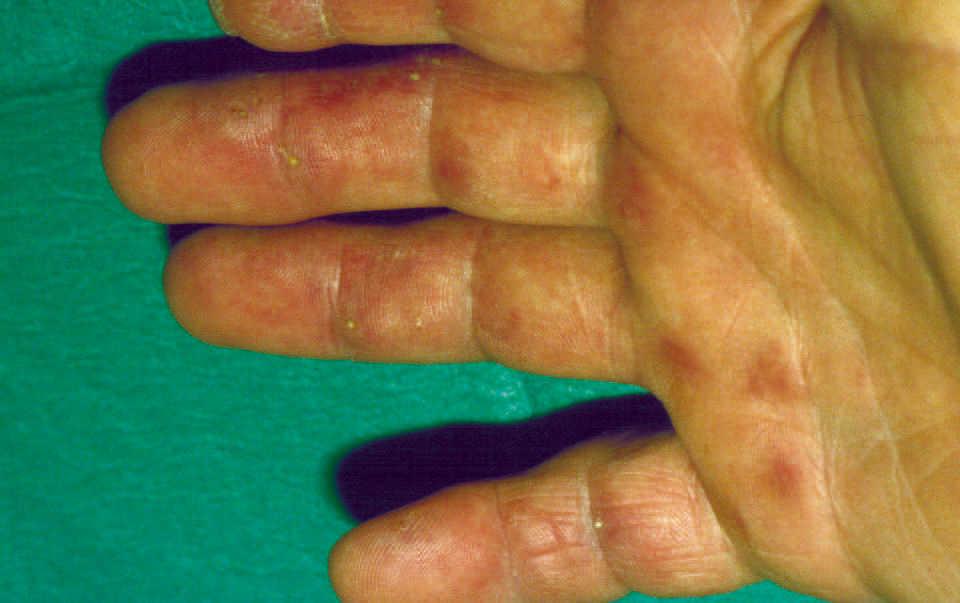
Figura 1. Espículas hiperqueratósicas en la cara palmar de los dedos.
Los estudios complementarios realizados (analítica de sangre y orina, proteinograma, hormonas tiroideas, estudio de autoanticuerpos y biomarcadores, serología de lúes, frotis de sangre periférica, endoscopia digestiva alta y baja, electromiografía, tomografía axial computarizada craneal y toraco-abdómino-pélvica, ecografía abdominal, radiografía simple de tórax, columna dorso-lumbar, caderas y articulaciones sacroilíacas) no mostraron hallazgos patológicos y descartaron una posible neoplasia oculta.
El examen histopatológico de la biopsia cutánea mostró columnas paraqueratósicas bien definidas con límites precisos con la epidermis ortoqueratósica normal adyacente, algunas de las cuales emergían de una capa granulosa moderadamente disminuida (fig. 2). La dermis superior mostraba un ligero infiltrado inflamatorio y algunos capilares dilatados. No se detectó vacuolización de queratinocitos ni disqueratosis.
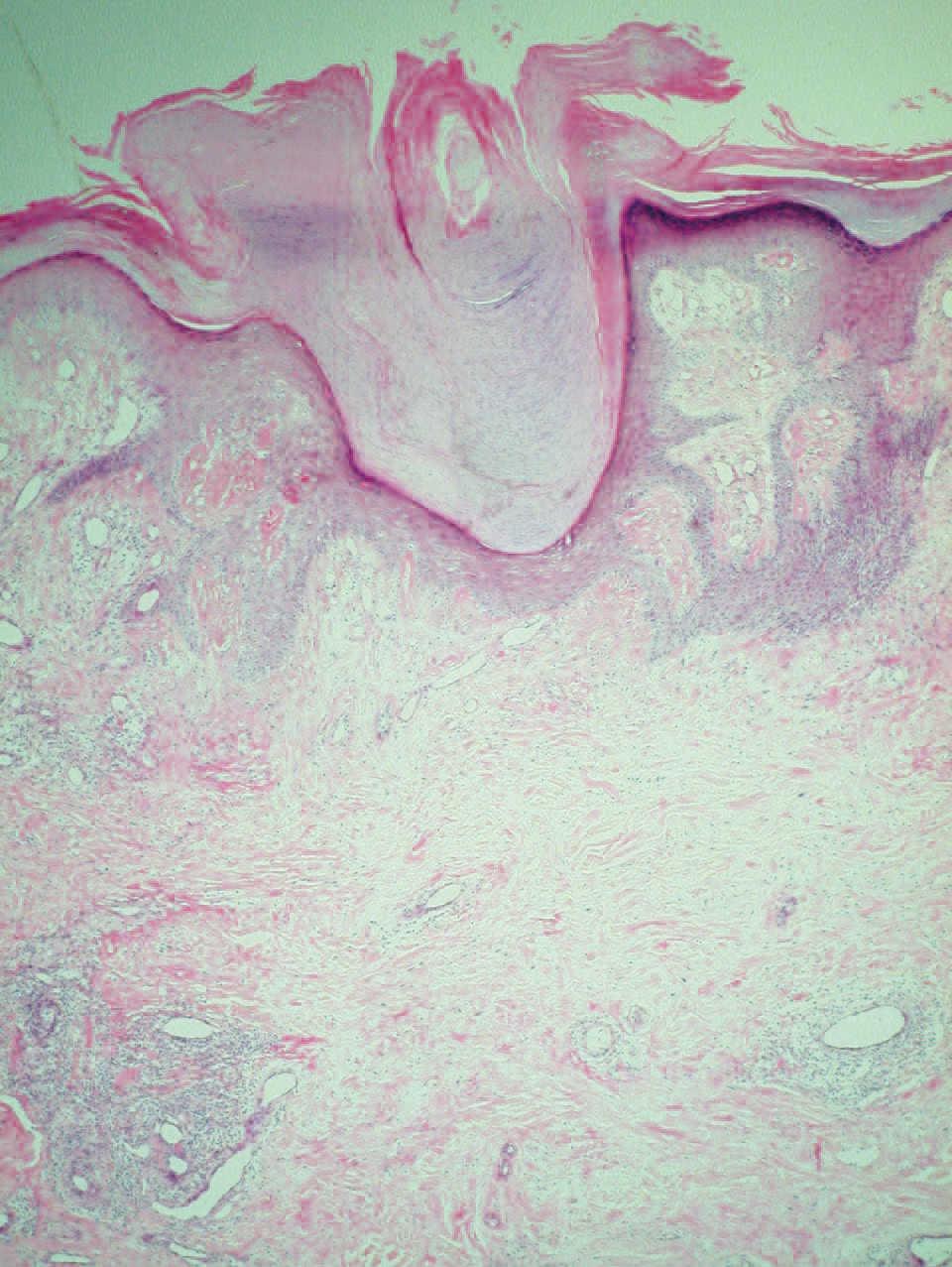
Figura 2. Columna paraqueratósica que asienta sobre una capa granular adelgazada. (Hematoxilina-eosina, 40.)
La aplicación de emolientes con urea tres veces al día produjo una mejoría de las lesiones, aunque muchas de ellas persistieron. En posteriores revisiones no se observaron nuevas lesiones.
Discusión
Desde que Goldstein describió el primer caso de lesiones digitiformes hiperortoqueratósicas en 19673, diferentes autores han comunicado casos similares con «espinas» hiperqueratósicas localizadas en palmas, plantas u otras superficies cutáneas, acuñando distintos términos en referencia a las mismas, como queratodermia punctata4, queratodermia poroqueratósica punctata5,6, poroqueratosis punctata7, poroqueratosis punctata palmaris et plantaris8, hiperqueratosis palmoplantar filiforme9,10, queratodermia espinosa1,11,12, hiperqueratosis palmar filiforme13 e hiperqueratosis digitada diminuta múltiple14. El hecho de que entidades similares recibiesen diferente denominación y el mismo término se emplease en referencia a distintos cuadros, explica la confusión que todavía existe en la actualidad en torno a esta dermatosis. Aunque algunos autores han realizado importantes intentos de agrupar y clasificar los casos descritos hasta el momento, la correcta denominación para esta entidad sigue siendo objeto de controversia.
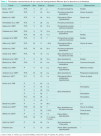
Las principales diferencias entre los casos descritos en la literatura son la localización de las lesiones y sus hallazgos histológicos, puesto que el aspecto clínico es muy similar en todos ellos. Estas observaciones condujeron a Zarour et al2 a dividir estas lesiones hiperqueratósicas en tres grupos (I, II y III) y dos subgrupos (a y b) en función de los hallazgos histológicos y de la localización de las lesiones, respectivamente (tabla 1). McGovern et al1 propusieron posteriormente una ligera modificación de la clasificación anterior e introdujeron el término «queratodermia espinosa» y los subtipos «paraqueratósica» y «ortoqueratósica». El caso que describimos reúne los criterios necesarios para ser clasificado como una HF tipo Ia, puesto que las lesiones se encontraban limitadas a las palmas de ambas manos y el principal hallazgo histológico fue la presencia de columnas paraqueratósicas bien definidas.
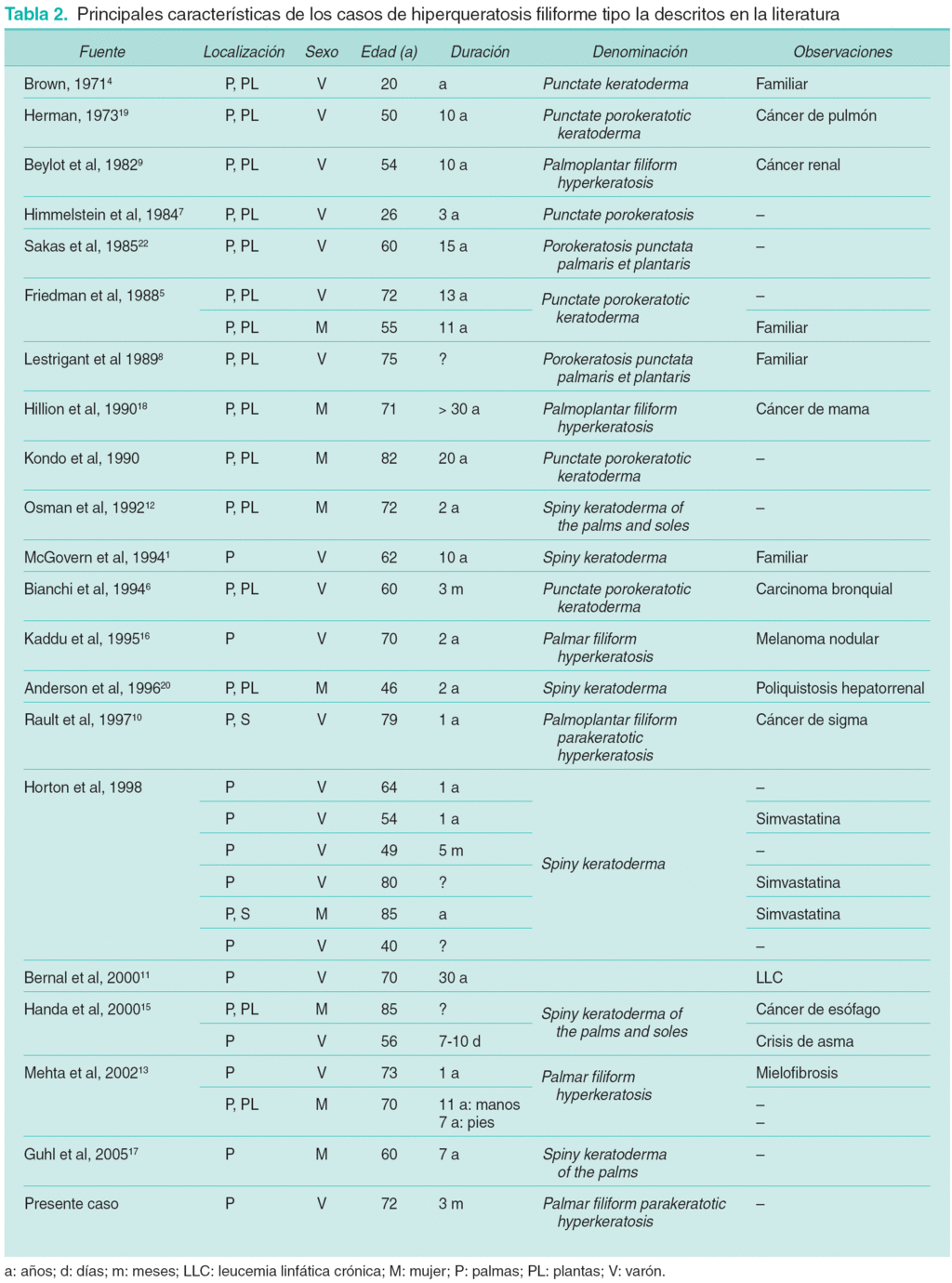
Mediante una búsqueda en la base de datos de PubMed empleando los descriptores «filiform hyperkeratosis», «palmar filiform hyperkeratosis» y «spiny keratoderma» encontramos 29 casos (incluyendo el presente) que podrían también ser clasificados como HF tipo Ia (tabla 2). Esta entidad parece mostrar una mayor incidencia en varones, puesto que de los 29 casos, 20 afectaban a varones y 9 a mujeres. Las lesiones aparecieron principalmente en pacientes de edad avanzada (media 62,48 años, rango 20-85 años). Las palmas y plantas se afectaban simultáneamente en 17 de los 29 casos y únicamente las palmas en 12 pacientes, incluyendo el nuestro1,11,13-17. Solamente en 9 pacientes se detectó un proceso tumoral subyacente: cáncer de mama18, de pulmón6,19, melanoma16, cáncer de sigma10, leucemia linfática crónica11, mielofibrosis13, cáncer renal9 y cáncer de esófago15. En otros casos las lesiones se asociaban con diversos procesos como poliquistosis renal10,20 o asma bronquial15.
Las lesiones hiperqueratósicas filiformes son habitualmente asintomáticas y miden 1-3 mm de altura, presentan una coloración blanquecino-amarillenta y emergen de una base no inflamatoria de la cual no se desprenden con facilidad. Sin embargo, en algunos casos las lesiones asentaban sobre áreas eritematosas (5, caso presente).
El principal hallazgo histológico de la HF paraqueratósica es una columna de células paraqueratósicas bien definida que recuerda a la lamela cornoide de las poroqueratosis. Para evitar confusión, Lestrigant y Berge8 acuñaron el término «paraqueratosis columnar» en referencia a este hallazgo. La columna paraqueratósica asienta generalmente sobre una capa granulosa reducida o incluso ausente y un estrato de Malpighio ligeramente adelgazado. Presenta límites muy bien definidos con respecto a la epidermis normal adyacente y ocasionalmente puede verse en relación con folículos pilosos5,16 y con el acrosiringio6,17,19, de forma similar al nevus ductal poroqueratótico ecrino21. Mehta et al13 describieron un caso que mostraba la coexistencia de columnas orto y paraqueratósicas. Raramente puede observarse vacuolización de queratinocitos22. En estos casos resulta más complicado rechazar firmemente la hipótesis que sugiere algún tipo de asociación entre esta entidad y la poroqueratosis, pero contrariamente a lo que sucede en ésta, en la HF paraqueratósica no se observa disqueratosis ni angulación en la columna. La dermis subyacente no muestra alteraciones significativas en la mayoría de los casos, sin embargo, en ocasiones puede observarse un ligero infiltrado inflamatorio15 y dilatación de capilares5,6.
No se han descrito casos de HF que hayan mejorado o se hayan resuelto de forma espontánea12. Numerosas alternativas terapéuticas han sido ensayadas con resultados variables, incluyendo retinoides tópicos20 y orales13, emolientes con ácido salicílico1,13, urea13, lactato amónico20, propilenglicol13 y 5-fluouracilo1,12.
Dada la variedad existente en cuanto a la presentación clínica y los hallazgos histopatológicos de las hiperqueratosis palmoplantares consideradas en conjunto, proponemos que más que entidades específicas sean consideradas un espectro clínico-patológico, cuya relevancia médica radica en su posible asociación (dudosa en algunos casos, pero claramente definida en otros) con neoplasias subyacentes.
A pesar de que la poroqueratosis y la HF paraqueratósica muestran claras diferencias clínicas e histopatológicas, ambas son trastornos de la queratinización que presentan similitudes morfológicas, genéticas y evolutivas y que pueden presentarse de forma concurrente con un proceso maligno o bien aparecer años antes o después del mismo. Por tanto, la realización de un estudio detallado en el momento del diagnóstico resulta especialmente importante en estos pacientes para descartar enfermedades sistémicas. Igualmente importante es el seguimiento del paciente a largo plazo, que puede evitar diagnósticos tardíos de neoplasias que hubieran podido recibir un tratamiento curativo.
En el momento actual, a la vista de los casos descritos en la literatura, parece adecuado descartar al menos neoplasias gastrointestinales, pulmonares y de mama. Los síntomas del paciente y los hallazgos patológicos extraídos de una exploración física rigurosa pueden aportar indicios valiosos para orientar la búsqueda en algunos casos.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Lidia Pérez-Pérez.
Servicio de Dermatología.
Facultad de Medicina.
San Francisco, s/n.
15782 Santiago de Compostela. A Coruña. España.
Correo electrónico: mejaime@usc.es
Aceptado el 3 de abril de 2007.


