

INTRODUCCION
Los fibromas escleróticos son neoplasias cutáneas benignas, generalmente solitarias, que aparecen en personas jóvenes como pequeñas pápulas o nódulos, blanquecinos o del color de la piel normal y de consistencia firme. Se localizan preferentemente, y por este orden de frecuencia, en la cara, las extremidades superiores e inferiores y el tronco (1). Cuando son múltiples pueden constituir un marcador cutáneo específico del síndrome de Cowden y su presencia ayuda en muchas ocasiones a un diagnóstico precoz de esta genodermatosis (2).
Estas lesiones se denominan fibromas escleróticos porque están constituidas en su mayor parte por una colágena con apariencia hialina, homogénea y eosinófila que se acompaña de un escaso número de fibroblastos dispuestos intersticialmente entre la colágena. Aunque se ha discutido mucho si el fibroma esclerótico, también denominado colagenoma estoriforme por algunos autores (3), corresponde o no a un dermatofibroma hipocelular de largo tiempo de evolución y con escasa actividad de sus células, un estudio reciente llevado a cabo por McCalmont ha demostrado que los fibroblastos presentes en la lesión sintetizan activamente colágeno tipo I y por tanto esta lesión no debe considerarse como un fósil de dermatofibroma (4).
Presentamos un ejemplo de fibroma esclerótico con células gigantes multinucleadas, una variante histopatológica poco frecuente de esta lesión.
DESCRIPCION DEL CASO
Varón de 35 años con un nódulo de consistencia firme de 1 cm de diámetro en la cara lateral externa de la falange distal del dedo anular de la mano derecha. La lesión había estado presente durante 3 años y mostraba un crecimiento lento. El diagnóstico clínico fue fibroma de las vainas tendinosas y la lesión fue extirpada quirúrgicamente. El resto de la exploración cutánea no demostró ninguna otra anomalía y no existía historia personal ni familiar de síndrome de Cowden.
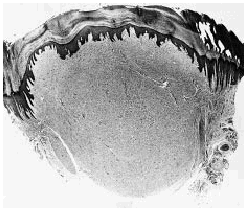
El estudio hispatológico demostró un nódulo dérmico bien delimitado, pero no encapsulado (Fig. 1), constituido por haces de colágena hialina y eosinófila dispuestos en algunas áreas con un patrón estoriforme y separados por grietas con abundante contenido de mucina y ausencia de tejido elástico. En la lesión se observaba escasa celularidad, pero existían numerosas células gigantes multinucleadas dispuestas entre los haces de colágena y especialmente abundantes en las áreas superficiales de la lesión (Fig. 2). Estas células multinucleadas mostraban un citoplasma amplio y eosinófilo y muchas de ellas tenían un contorno poligonal o estrellado con bordes angulosos. Los núcleos de estas células multinucleadas eran monoformos y no se observaban atipias ni figuras de mitosis. Desde el punto de vista inmunohistoquímico las células multinucleadas mostraban positividad para la vimentina, pero eran negativas para la proteína S-100, las citoqueratinas AE1/AE3, la actina muscular específica y el factor XIIIa.
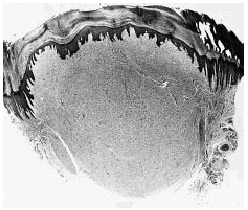
FIG. 1.--Nódulo dérmico hipocelular bien delimitado, aunque no encapsulado.

FIG. 2.--La lesión está constituida por abundantes haces de colá-geno hialinizado y escaso número de células. Las células gigantes multinucleadas son especialmente abundantes en las áreas superficiales de la lesión.
DISCUSION
El fibroma esclerótico es una lesión benigna, descrita originalmente en 1972 por Weary y cols. (5) en la lengua de un paciente con síndrome de Cowden, una genodermatosis de herencia autosómica dominante que asocia lesiones mucocutáneas consistentes en tricolemomas faciales múltiples, pápulas verrugosas en mucosa bucal, pápulas translúcidas y queratósicas en los pulpejos de los dedos y frecuente desarrollo de carcinomas mamarios y tiroideos. Después de su descripción original otros autores han publicado ejemplos de fibromas escleróticos en pacientes con síndrome de Cowden localizados en la cara, las palmas y las plantas (6, 7).
Los primeros autores que describieron esta lesión aislada, es decir, sin asociación con síndrome de Cowden, fueron Rapini y Golitz (1) en 1989. Estos autores publicaron una serie de 11 pacientes con fibromas escleróticos solitarios, localizados en diversas áreas de la superficie corporal y que no presentaban ninguna de las anomalías del síndrome de Cowden.
Requena y cols. en 1992 describieron un paciente con síndrome de Cowden y fibromas escleróticos múltiples. Estos autores revisaron la literatura al respecto y llegaron a la conclusión de que la presencia de fibromas escleróticos múltiples constituía un marcador cutáneo de síndrome de Cowden que puede ayudar al diagnóstico precoz de esta genodermatosis. En contraste, los pacientes con fibromas escleróticos solitarios no suelen padecer síndrome de Cowden (2).
Desde el punto de vista hispatológico la lesión es muy característica. Consiste en un fibroma mayoritariamente constituido por gruesos haces de colágena y escaso número de fibroblastos. En algunas áreas los haces de colágena muestran una disposición estoriforme o en hojas de cebolla.
En nuestra revisión de la literatura sólo hemos encontrado el artículo de Rudolph y cols. (8) sobre fibroma esclerótico con células gigantes multinucleadas. Estos autores describieron cinco ejemplos de fibroma esclerótico solitario con abundantes células gigantes multinucleadas. Nuestro caso es idéntico a los descritos por ellos.
El diagnóstico diferencial del fibroma esclerótico con células gigantes multinucleadas debe plantearse con el nevo de Spitz intradérmico, con el angiohistiocitoma de células gigantes multinucleadas y el fibroma pleomórfico. El diagnóstico de nevo de Spitz intradérmico en nuestro caso se excluyó fácilmente por la negatividad de la proteína S-100 en las células gigantes multinucleadas. El angiohistiocitoma de células gigantes multinucleadas es una rara entidad que habitualmente consiste en múltiples pápulas de apariencia angiomatosa agrupadas en una región anatómica o salpicadas por las extremidades (9, 10). Aunque el angiohistiocitoma de células gigantes multinucleadas muestra el mismo tipo de células gigantes multinucleadas, con citoplasma poligonal y bordes angulosos, existe además una llamativa proliferación de vasos sanguíneos de tipo capilar como parte integrante de la lesión y no muestra el grado de colagenización del fibroma esclerótico. Respecto al fibroma pleomórfico, descrito originalmente por Kamilo y cols. (11), también puede mostrar células gigantes multinucleadas similares a las del fibroma esclerótico de células gigantes multinucleadas, pero estas células multinucleadas del fibroma pleomórfico muestran núcleos atípicos e hipercromáticos y la lesión carece de la colagenización y el patrón estoriforme del colágeno que caracterizan al fibroma esclerótico.


