






Métodos moleculares de estudio en Farmacogenética. Tecnología de los microarrays de ADN
Desde la década de los ochenta se ha venido analizando la estructura de los genes y su expresión de manera individualizada mediante técnicas como Southern Blot y Northern Blot (hibridación de una sonda con ADN y ARN respectivamente). Con ellas se llevaba a cabo el análisis individualizado de un gen o de un grupo reducido de genes cada vez. El problema es que para encontrar asociaciones entre un gen y una enfermedad había que probar cientos o miles de posibilidades en lo que sería un proceso lento, caro, pesado y que, con frecuencia, no conducía a nada. Estudiar la expresión del genoma completo de gen en gen sería como vaciar el océano con una cucharilla. Sin embargo, las nuevas técnicas de microarrays permiten analizar miles de genes en un único experimento 1. En un futuro se convertirán en técnicas de rutina. En la actualidad la técnica más empleada para llevar a cabo estos estudios se realiza mediante la tecnología de los microarrays o chips de ADN 2.
Los orígenes de la tecnología de los microarrays o chips de ADN se remonta al inicio de los años noventa 3. Desde entonces su uso en investigación biomédica ha aumentado de forma espectacular en los últimos años, y abre unas enormes expectativas para el futuro. La técnica se fundamenta en la complementariedad de las dos cadenas de los ácidos nucleicos (permite detectar secuencias específicas de ARN o ADN basándose en su capacidad de combinarse específicamente a secuencias complementarias de una sonda de ADN) 4.
El microarray es un soporte al que se han unido fragmentos de genes, oligonucleótidos (fragmentos cortos de ADN), o productos procedentes de la reacción en cadena de la polimerasa (PCR), por lo que el «encuentro» con material procedente del tejido del enfermo permite identificar, gracias a la complementariedad, aquellos genes que están presentes. Con ello se consigue la identificación de genes presentes en los tejidos normales o enfermos comparando la carga y la expresividad de cada una de las muestras. Permite situar en los escasos centímetros cuadrados de un vidrio especial, en posiciones definidas y concretas, hasta cientos de miles de fragmentos de ADN con una secuencia concreta (en su conjunto representan varias veces los genes del genoma). Se fabrican empleando la misma tecnología que se utiliza para elaborar los chips semiconductores, pero utilizando millones de cadenas de ADN colocadas verticalmente en un chip de cristal o silicona. Para ello se utiliza la llamada combinatorial chemistry. Se llegan a sintetizar hasta 1,3 millones de sondas de oligonucleótidos en cada array (cada oligonucleótido se coloca en un área específica del array llamada «celdilla de la sonda» o probe cell. Cada probe cell contiene cientos de miles de millones de copias de un determinado oligonucleótido).
El diseño y la fabricación de los microarrays de ADN dependen en primera instancia del ácido nucleico que posteriormente se vaya a estudiar. Para su construcción es esencial la caracterización total o parcial del genoma. La secuenciación de miles de clones procedentes de genotecas de ADN genómico y de ADNc permite identificar los distintos genes. A partir de esa información, almacenada en grandes bases de datos como GeneBank (http://www.ncbi. nih.gov) o EMBL (http://www.ebi.ac.uk),
se identifican clones representativos de cada uno de los genes. El ADN presente en cada uno de ellos es amplificado mediante PCR y purificado, para ser posteriormente depositado, mediante robots de alta precisión, sobre soportes de vidrio que constituyen los chips de ADN. Alternativamente, a partir de la secuencia de los genes o de los ADNc se diseñan oligonucleótidos de pequeño tamaño (25-70 residuos) que específicamente hibridan con cada uno de ellos. Los oligonucleótidos son previamente sintetizados antes de inmovilizarlos sobre los soportes, o bien sintetizados in situ sobre el chip. De esta última tecnología los diferentes proveedores emplean métodos también diferentes: Affymetrix® (www.affymetrix.com), la fotolitografía; Agilent® (www.agilent.com), inyección de precursores fosforoamiditos; Roche® (www.roche-applied-science.com/sis/matrix array/), activación de precursores por campo eléctrico.
Los desarrollos futuros ofrecerán chips específicos de enfermedad o de respuesta a fármacos, presumiblemente más baratos y de un manejo más sencillo que los dispositivos actuales, de tal manera que puedan universalizarse en los sistemas de salud tanto públicos como privados. Aunque todavía existe cierta controversia sobre la sensibilidad y especificidad de estas técnicas 5, en el momento presente están empezando a ser comercializados.
Esta técnica permite analizar el comportamiento, es decir, la inducción o represión de todos los genes que se expresan en un tejido como consecuencia, por ejemplo, de una enfermedad o un tratamiento. Detecta pérdidas o ganancias cromosómicas, polimorfismos, mutaciones y cambios en los niveles de expresión de los genes. Estos resultados nos pueden permitir establecer si una persona tiene un «patrón» o «perfil» de enfermedad, es susceptible de desarrollarla, o puede responder o no a un tratamiento.
Se distinguen tres tipos de microarrays:
1.Microarrays para determinar la expresión génica: detectan secuencias específicas de ARN.
2.Microarrays para determinar el genotipo: detectan secuencias específicas de ADN.
3.Microarrays para determinar la resecuenciación.
Dado que la expresión génica, más que la propia dotación génica, y los polimorfismos existentes en ellos es lo que explica y condiciona las diferencias de los individuos en la respuesta a los tratamientos, objeto de esta revisión, nos centraremos fundamentalmente en los dos primeros tipos de estudios con microarrays.
Análisis de la expresión génica
No todos los genes de los que el individuo es portador se expresan; sólo lo hace una fracción de ellos. La expresión génica es el mecanismo por el que la información contenida en el ADN da lugar a ARN mensajero (ARNm) y, finalmente, este proceso se traduce en la síntesis de una proteína. La expresión génica permite a la célula adaptarse y responder a los estímulos procedentes de otras células o del ambiente. La importancia que tiene conocer cuáles son los genes «activos» es trascendental para explicar el perfil genético en cada situación. Un estudio fundamental con referencia a la determinación de la expresión génica mediante tecnología de microarrays de ADN fue llevado a cabo por Lockhart et al en 1996 6. En él, oligonucleótidos de ADN permitieron identificar unas pocas moléculas de ARNm por célula. En aquella época, los microarrays de ADN podían detectar la expresión génica de unos 1.000 genes. En la actualidad, los microarrays llevan sondas de todos los genes conocidos hasta la actualidad (varios miles).
La expresión génica se estudia midiendo la cantidad de copias de ARN que produce un gen (gen activo). No detecta, por tanto, aquellos genes que, existiendo, permanecen inactivos. Los análisis de expresión génica con microarrays con frecuencia se realizan a ciegas inicialmente. Se estudia un panel amplio de genes y posteriormente se analizan los resultados y su posible influencia en la enfermedad o en la respuesta terapéutica. Permiten al investigador comenzar sin una hipótesis concreta. Pueden medir la expresión génica de cada uno de los genes conocidos en el genoma humano completo, incluso de genes de los que se desconoce su función. Simplemente comparando los patrones de expresión génica (por ejemplo pacientes con psoriasis respondedores a ciclosporina y no respondedores) se pueden detectar diferencias en estos patrones. En el caso de que se demuestre una diferencia de patrones en la expresión de los genes tiene sentido iniciar una investigación dirigida.
Los microarrays para determinar la expresión génica se basan en la atracción química natural (llamada hibridación) entre el ADN (en el array) y las moléculas de ARN (en la muestra de estudio) para determinar qué secuencias de ARN se están expresando en una determinada muestra y su nivel de expresión a partir de un determinado gen (qué ARN y la cantidad de ARN que se está produciendo). Cuando una cadena de ADN fabrica una cadena de ARN, las dos cadenas son complementarias porque los pares de bases se corresponden (A, T, G y C en la cadena de ADN, con U, A, C y G en la cadena de ARN respectivamente). Si las bases no son complementarias no se unirán el ADN y el ARN (una sola base que no se complementa es capaz de impedir que se unan las dos cadenas).
Existen dos tipos de microarrays para determinar la expresión génica. En la actualidad ambas tecnologías tienen una distribución similar en los laboratorios de investigación del mundo:
1.Microarrays de oligonucleótidos de ADN. Se inmovilizan pequeños fragmentos de ADN sintetizados químicamente (oligonucleótidos) específicos para cada gen expresado. Actualmente por su homogeneidad, reproducibilidad y robustez son los más usados. Existen varios proveedores: Affymetrix® (es la principal compañía proveedora de este tipo de microarrays. Posee chips de alta densidad que con sus más de 500.000 oligonucleótidos permiten analizar simultáneamente la expresión de más de 20.000 genes) (fig. 1), Agilent® y Roche®.

Figura 1. Microarray de oligonucleótidos de ADN (Affymetrix, Santa Clara, EE.UU.).
2.Microarrays de ADNc. Emplean sondas de ADN con aproximadamente 600-2.000 bases de longitud, en lugar de las 25, sobre una base de cristal, nitrocelulosa o nylon. Se inmovilizan fragmentos de ADNc procedentes de colecciones de clones (genotecas).
Metodología en la técnica de microarrays de oligonucleótidos
La metodología en la técnica de microarrays de oligonucleótidos de expresión génica incluye las siguientes fases:
1.Extracción y preparación del ARN a partir de una muestra (sangre o tejido). Se obtiene el ARN total a partir de los leucocitos en las muestras de sangre y/o del tejido tras un proceso de machacado o molido (grinding) en estas últimas.
2.Amplificación y secuenciación del ARN. Con el fin de facilitar la hibridación, se realiza una amplificación (millones de copias) del ARN mediante PCR. Posteriormente se fragmentan las cadenas de ARN en millones de secciones. Además se añaden moléculas de biotina que actúan como pegamento para moléculas fluorescentes.
3.Identificación del ARN. La determinación de la expresión de los genes candidatos se realiza mediante microarrays de oligonucleótidos de alta densidad. Este microarray examina prácticamente cada gen del genoma humano. Contiene más de 45.000 fragmentos (probe sets) que corresponden a 33.000 genes conocidos y 6.000 expressed sequence tags (EST), es decir, aproximadamente todo el genoma humano. Ello permite la identificación de aquellos genes, conocidos o desconocidos previamente, que estén expresados, y también la cuantificación del nivel de expresión de cada uno de ellos.
En estos microarrays las sondas de ADN van colocadas en la superficie de una base de cristal. Cada sonda contiene generalmente tan sólo 25 bases de longitud que suponen una pequeña sección, pero muy representativa de un gen completo con muchísimas más bases. Si una cadena de ARN se combina con estas 25 bases, se podrá saber de qué gen concreto se trata. El microchip es tan preciso que puede detectar incluso una sola molécula de ARN específico de un gen dentro de 100.000 ARN diferentes que también puedan estar presentes en la muestra (sensibilidad de detección de aproximadamente 1:100.000).
La superficie de este microarray es como un tablero de damas de 1,25 × 1,25 cm. La superficie total contiene hasta 1,3 millones de cuadrículas. Cada cuadrícula (feature) mide 11 × 11 micrometros y contiene millones de copias de cada sonda de un mismo ADN (fig. 2). Las sondas se van apilando unas junto a otras por un mecanismo de síntesis fotolitográfica.
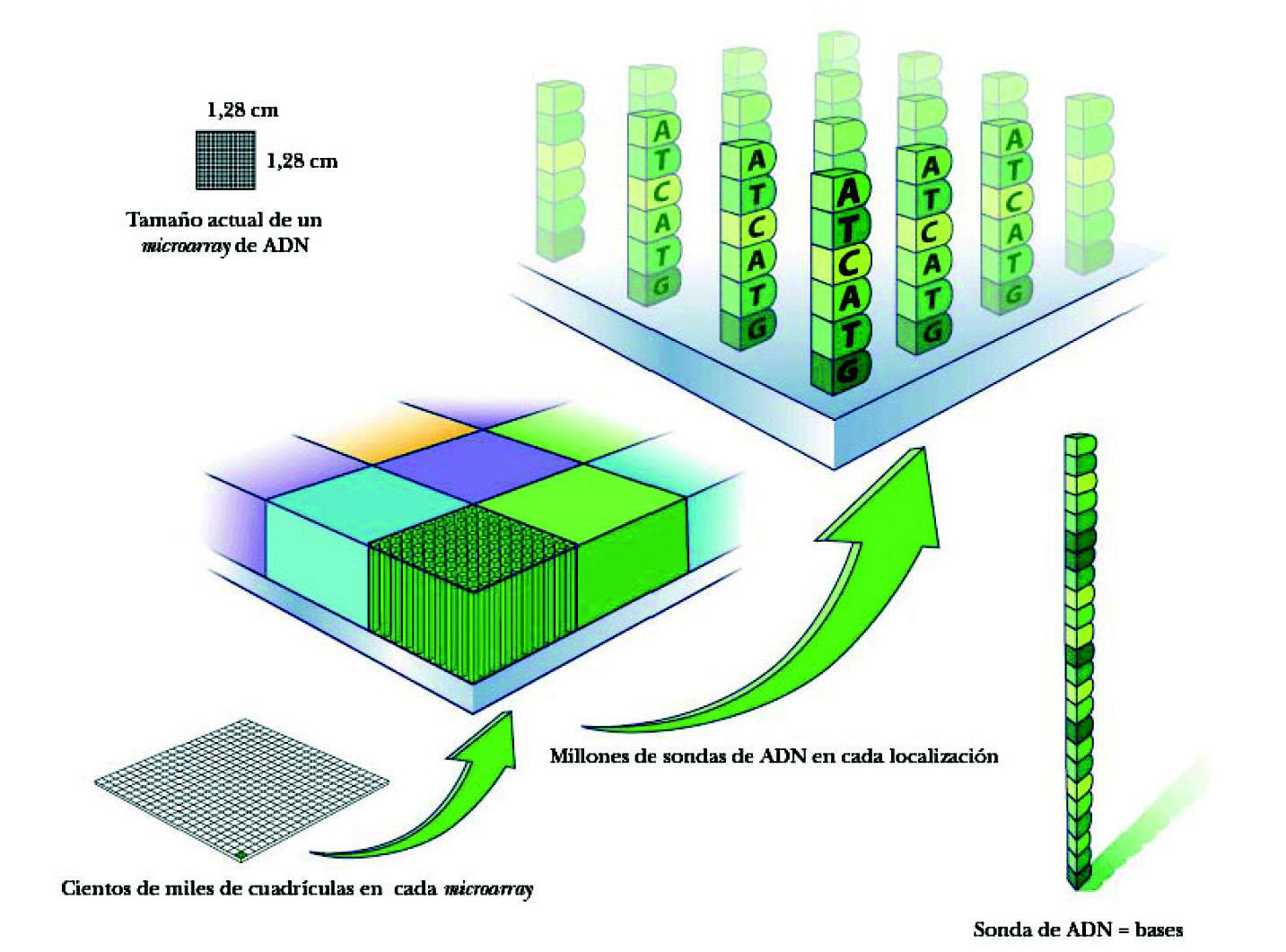
Figura 2. Composición de un microarray de ADN.
El proceso de identificación del ARN consta de los siguientes pasos: a) colocación del ARN en el microarray. El ARN extraído de la muestra y multiplicado se deposita en el microchip. Se deja 14-16 horas para permitir que la hibridación tenga lugar (para que se complementen las bases de ARN de la muestra y ADN de las sondas del microarray). Cada cadena de ARN de los genes que se han expresado está nadando entre las sondas de ADN, buscando su complemento perfecto. Aunque desgraciadamente la mayor parte de ellas no encuentra su combinación en el array, algunas sí lo hacen y quedan como adheridas a la sonda de ADN (fig. 3); b) aclarado del microarray. Posteriormente se aclara el microarray para eliminar el ARN no combinado. Por el contrario permanece el ARN hibridado a la sonda de ADN, a su vez unida a la molécula de biotina, y c) observación del ARN hibridado. Para poder ver el ARN combinado con la sonda de ADN se utiliza una molécula fluorescente que se une a la biotina. Tras un lavado, las moléculas fluorescentes no combinadas se eliminan de manera que sólo quedan las moléculas fluorescentes unidas a biotina, expresión de las cadenas de ARN unidas a las sondas de ADN. Por tanto, la detección de luz fluorescente será síntoma de hibridación y, por ello, de detección de una secuencia específica determinada. La lectura de la fluorescencia se realiza mediante un láser escáner. La intensidad de las radiaciones informará de la cantidad relativa de cada secuencia de la muestra. Dependiendo de la cantidad de ARN hibridado, brillará más o menos. Si un gen está muy expresado y existen muchas moléculas de ARN, brillará mucho. Por ello, según la intensidad del brillo se puede cuantificar la cantidad de ARN. A su vez, valorando todas las cuadrículas se podrán evaluar todos los varios miles de genes presentes en el array. En síntesis, conociendo la identidad de una sonda de oligonucleótido (y la de su gen correspondiente) por su localización en el microarray podremos identificar el gen expresado y por la intensidad de fluorescencia podremos determinar su nivel de expresión (fig. 4).
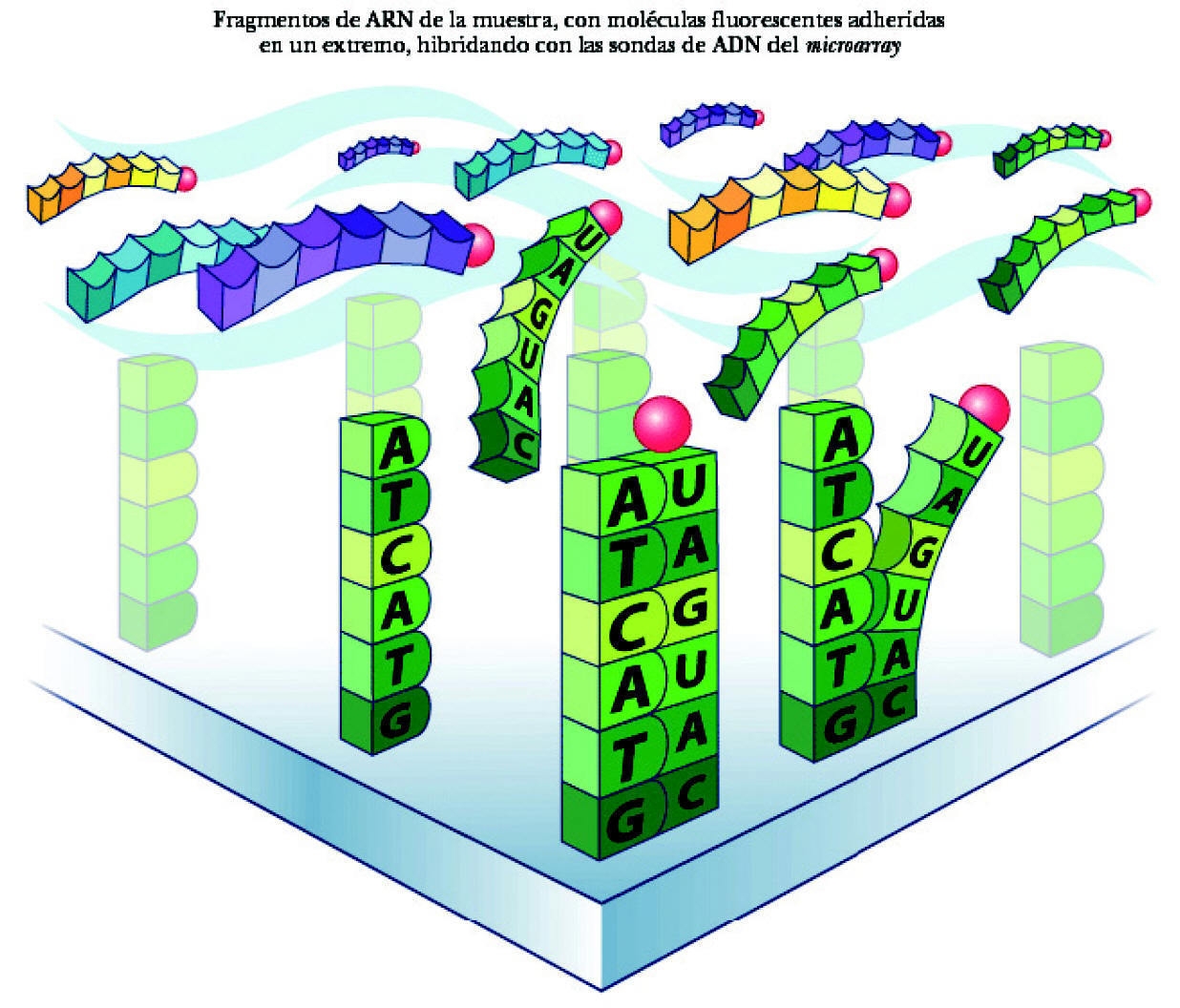
Figura 3. Hibridación del ARN.
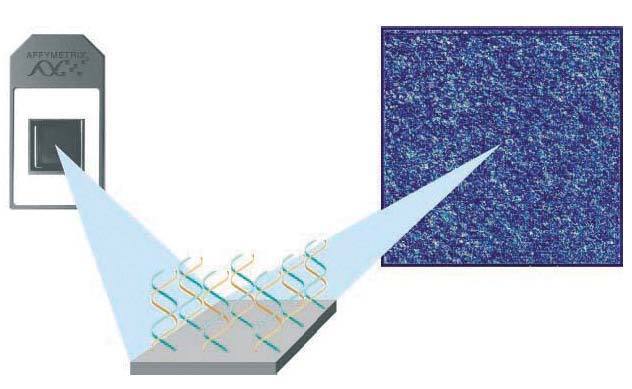
Figura 4. Resultado del ARN hibridado.
Para poder comparar entre unas y otras muestras se construye un «mapa de calor» (heat map). Según la intensidad de la expresión, tras aplicar el láser, los features mostrarán unos u otros colores. Son representaciones gráficas que colorean la expresión génica. Solo se colorearán intensamente aquellos genes que estén muy expresados. Estos mapas se pueden grabar, registrar y almacenar, para así poder ser valorados pasado un tiempo. La imagen obtenida de esta manera es analizada informáticamente (fig. 5).
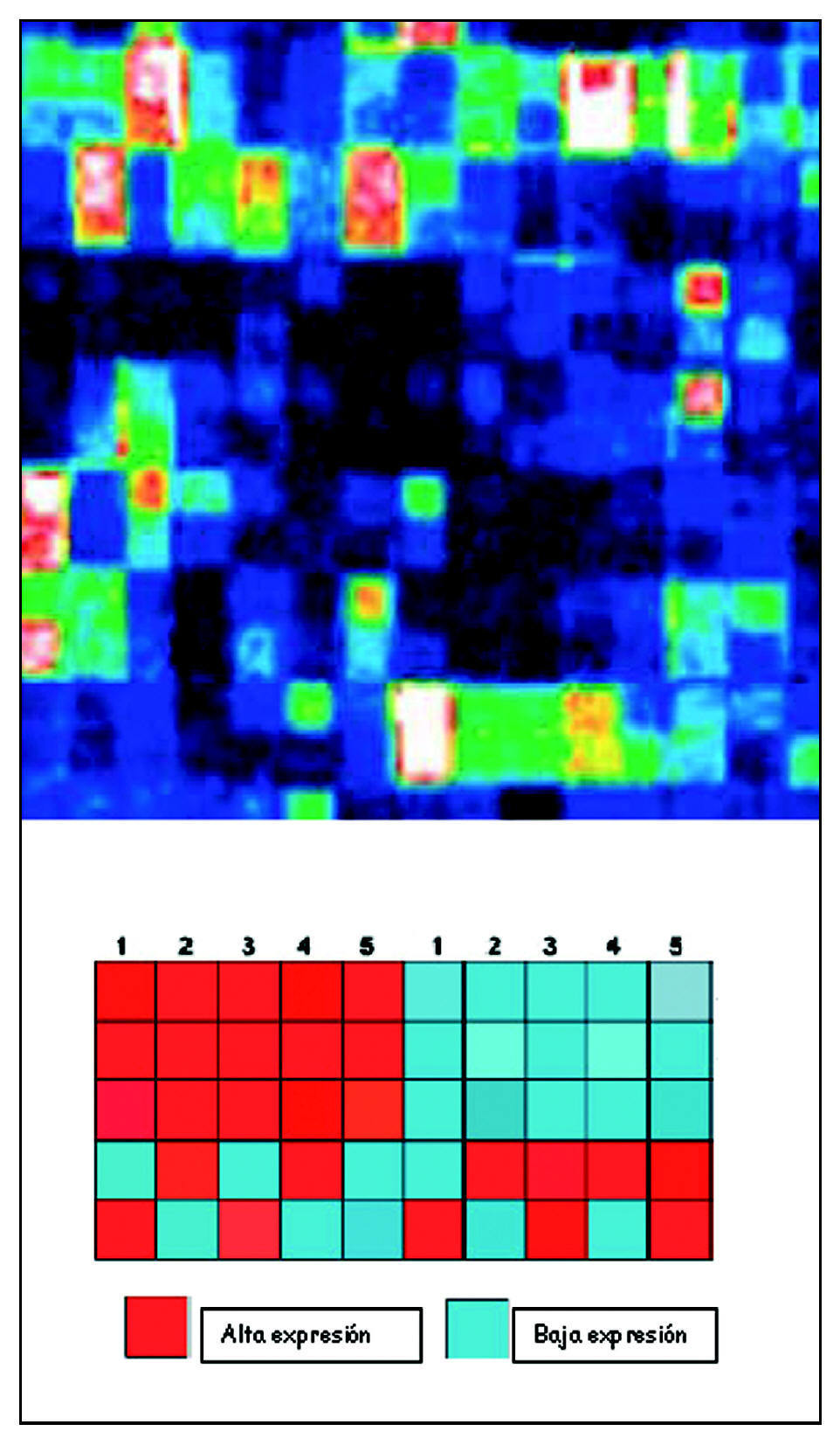
Figura 5. Mapa de calor.
Utilidades de la técnica de microarrays de oligonucleótidos
Las utilidades de esta tecnología son:
1.Clasificación de enfermedades. Si comparamos la expresión génica entre diferentes pacientes con una determinada enfermedad, las diferencias entre ellos quizás puedan indicarnos subtipos de la misma. Todos ellos tienen la misma enfermedad y probablemente compartirán la expresión de varios genes, pero también podrán expresar otros relacionados con la enfermedad que aportarán matices.
2.Conocimientos de los mecanismos fisiopatológicos de las enfermedades. Una vez determinada la expresión de un gen, si se conoce su función, se puede obtener mucha información sobre los mecanismos patogénicos de una determinada enfermedad.
3.Investigación de nuevos fármacos. En función del apartado anterior, el conocimiento de mecanismos patogénicos de una enfermedad permite la investigación de nuevos fármacos que tengan por diana aquellos mecanismos en los cuales interviene ese gen, pero también es útil para determinar su toxicidad, no sólo su eficacia.
4.Predicción de la respuesta terapéutica de los pacientes a los fármacos y optimización de su utilización: terapia individualizada. Ya ha sido comentado previamente que los pacientes con una determinada enfermedad común responden de forma diferente a los mismos fármacos. Si comparamos la expresión génica de un paciente antes y después de recibir un fármaco, veremos las diferencias que existen. Si comprobamos que los pacientes respondedores tienen un patrón característico, frente a los no respondedores, podremos predecir qué pacientes se beneficiarán de un fármaco y cuáles no. De esta forma valoramos eficacia. Si realizamos lo anterior con diferentes dosificaciones de un fármaco, podremos también comprobar qué dosis son necesarias para determinados patrones de pacientes (por ejemplo aquellos con determinado patrón de expresión génica no son respondedores a la dosis habitual, pero sí lo son a dosis más altas). De esta manera podremos individualizar y ajustar la dosis necesaria. Y lo mismo podríamos decir de la toxicidad; podremos predecir qué pacientes sufrirán efectos secundarios y cuáles no.
Análisis del genotipo
Aunque la mayor parte de la secuencia del genoma es homogénea, alrededor de uno de cada 100-1.500 nucleótidos es polimórfico, es decir, tiene una base en un cromosoma distinta a la de otro cromosoma. Estas diferencias, llamadas polimorfismos, tienen consecuencias sobre la expresión de la proteína o sobre su estructura lo que hace que, en la práctica, sólo los gemelos monocigóticos tengan una carga genética altamente semejante, mientras que hay diferencias muy notables entre los individuos de la especie humana. Cada persona hereda dos copias de cada gen, una de cada uno de sus padres. Estas copias pueden ser idénticas o diferentes. El conjunto de estas diferencias heredables constituye la variación genética entre individuos. Muy pequeñas diferencias en la secuencia de ADN pueden tener grandes efectos en la salud y la enfermedad. Un gen que funciona correctamente en una persona puede hacerlo de forma reducida o incluso no funcionar en otra. Los polimorfismos genéticos pueden ser definidos cuando existen variaciones monogénicas en la población normal con una frecuencia mayor del 1 % 7. Los polimorfismos pueden ser:
1.Mutaciones importantes del ADN, con repercusiones muy notables como: a) delecciones (eliminación de una o más bases o segmentos de la secuencia de ADN). Generalmente tiene una repercusión negativa importante en el gen afectado y en la proteína que es codificada por él; b) inserciones (se añade una base o una sección de bases, extras, a la secuencia de ADN). Puede tener lugar en la zona de codificación del gen o no. Generalmente tienen efectos extremadamente negativos sobre las proteínas codificadas.
2.Pequeñas mutaciones, con menor repercusión. Incluyen los polimorfismos de un único nucleótido (SNP), el tipo más frecuente de polimorfismo.
Los SNP son variaciones, homo o heterocigotos, de un solo par de bases en la secuencia de ADN del genoma humano. Son la forma más simple y frecuente de polimorfismos entre individuos. Constituyen una de las herramientas más poderosas para el análisis de genomas. La mayor parte de los SNP se encuentran fuera de las regiones codificadoras o promotoras de los genes. Sin embargo, tanto los que se encuentran dentro como fuera de esas zonas pueden tener influencia en la transcripción de los genes. Las características más importantes de los SNP, que hacen de ellos unos marcadores genéticos ideales en la búsqueda de genes de susceptibilidad de una enfermedad o genes que determinan la respuesta a un fármaco son: simplicidad, amplia distribución, estabilidad y alta frecuencia a lo largo del genoma (se estima que tienen una incidencia de aproximadamente un SNP por cada 1.000 pares de bases) 8.
Se han identificado más de 1,4 millones de SNP en la secuenciación inicial del genoma humano (con más de 60.000 de ellos en las zonas de codificación de los genes) 9, y se estima que el genoma humano contiene unos 10 millones de SNP. En la actualidad, el Consorcio SNP (grupo sin ánimo de lucro formado por compañías farmacéuticas, compañías procesadoras de información, instituciones académicas y fundaciones de investigación) ha determinado un mapa de alta densidad con varios millones de SNP (http://snp.cshl.org). Gracias a este mapa, actualmente miles de polimorfismos se pueden identificar y ordenar de forma precisa.
En el pasado, y todavía en el presente, para conseguir los objetivos de la farmacogenética se seguían, y se siguen, los pasos detallados a continuación: a) se analiza la distribución de la población para la variación en estudio utilizando una sonda válida para detectar el polimorfismo fenotípico (que afecta a > 1 % de la población); b) se identifica el gen responsable y sus variantes; c) se realizan estudios familiares y de gemelos para confirmar sus características genéticas (herencia dominante, recesiva, mendeliana, etc.); d) se desarrollan ensayos genéticos para las distintas variantes del ADN; e) se realiza la correlación entre el genotipo y el fenotipo, y f) se aplica a la práctica clínica.
De hecho, la mayoría de los estudios de farmacogenética publicados están basados en correlacionar la respuesta del paciente con las variaciones en los genes involucrados en el mecanismo de acción del fármaco o de su metabolismo con el fin de determinar el «gen candidato». Por el contrario, en la última década, los avances en la tecnología de secuenciación molecular permiten, además, una investigación sistemática para encontrar variaciones funcionales significativas en las secuencias de ADN de los genes que influyen en los efectos de diversos fármacos, para después estudiar su repercusión fenotípica. Esto se puede hacer hoy en día gracias nuevamente a los microchips de ADN para detectar SNP 8-10. No es imprescindible conocer de antemano el gen responsable, sino que se puede llegar a él desde el análisis de las diferencias entre respondedores y no respondedores. Se basa en la complementariedad, en la atracción de una cadena de ADN con otra cadena de ADN (y en la combinación de pares de bases: ATCG con TAGC respectivamente). Para determinar el genotipo se emplean chips de oligonucleótidos de manera casi exclusiva. Affymetrix® es también el principal proveedor, con microchips que permiten analizar miles de SNP conocidos en un único experimento (actualmente se están desarrollando microarrays de hasta 100.000 SNP). Dado que el Proyecto Genoma Humano ha permitido secuenciar gran parte del mismo, en el microarray está reflejado tal conocimiento y contiene las secuencias de ADN (secuencias cortas pero representativas de los genes del genoma), de manera que se podrá detectar el genotipo completo (incluyendo los SNP) del individuo que aporte la muestra.
Determinar el genotipo de un individuo sólo tiene que realizarse una vez para un determinado gen, porque, con excepción de raras mutaciones, no suele cambiar (a diferencia del análisis de expresión del ARN, en el que la expresión génica de los diferentes tipos celulares y, por tanto, la producción de ARN es variable) y además se puede realizar en cualquier tejido o fluido, ya que el ADN es común a todas las células del organismo.
Metodología de la técnica de microarrays para la detección de polimorfismos de un único nucleótido
La metodología en la técnica de microarrays para detectar SNP es similar a la descrita para la expresión génica:
1.Extracción y multiplicación del ADN a partir de una muestra (sangre o tejido).
2.Amplificación y secuenciación del ADN. Con el fin de facilitar la detección de los SNP se realiza una amplificación (millones de copias) de cada fragmento del ADN que contiene estos polimorfismos de la muestra mediante PCR. Posteriormente se secuencia el ADN.
3.Identificación del genotipo de ADN. La muestra obtenida anteriormente, con el ADN, se introduce en un microarray para la detección de SNP. La estructura del microarray es similar a la empleada en el análisis de ARN, con sondas de oligonucleótidos de ADN con 25 bases dispuestas apiladas unas con otras en cientos de miles de cuadrículas (cada cuadrícula con un solo tipo de sonda de ADN). Incluye la mayoría de los genes del genoma humano. Permite analizar miles de SNP conocidos en un único experimento. El proceso de identificación del ADN sigue las mismas fases que para el ARN:
a)Colocación del ADN en el microarray.
b)Aclarado del microarray (para seleccionar el ADN hibridado).
c)Observación del ADN hibridado (fig. 6).
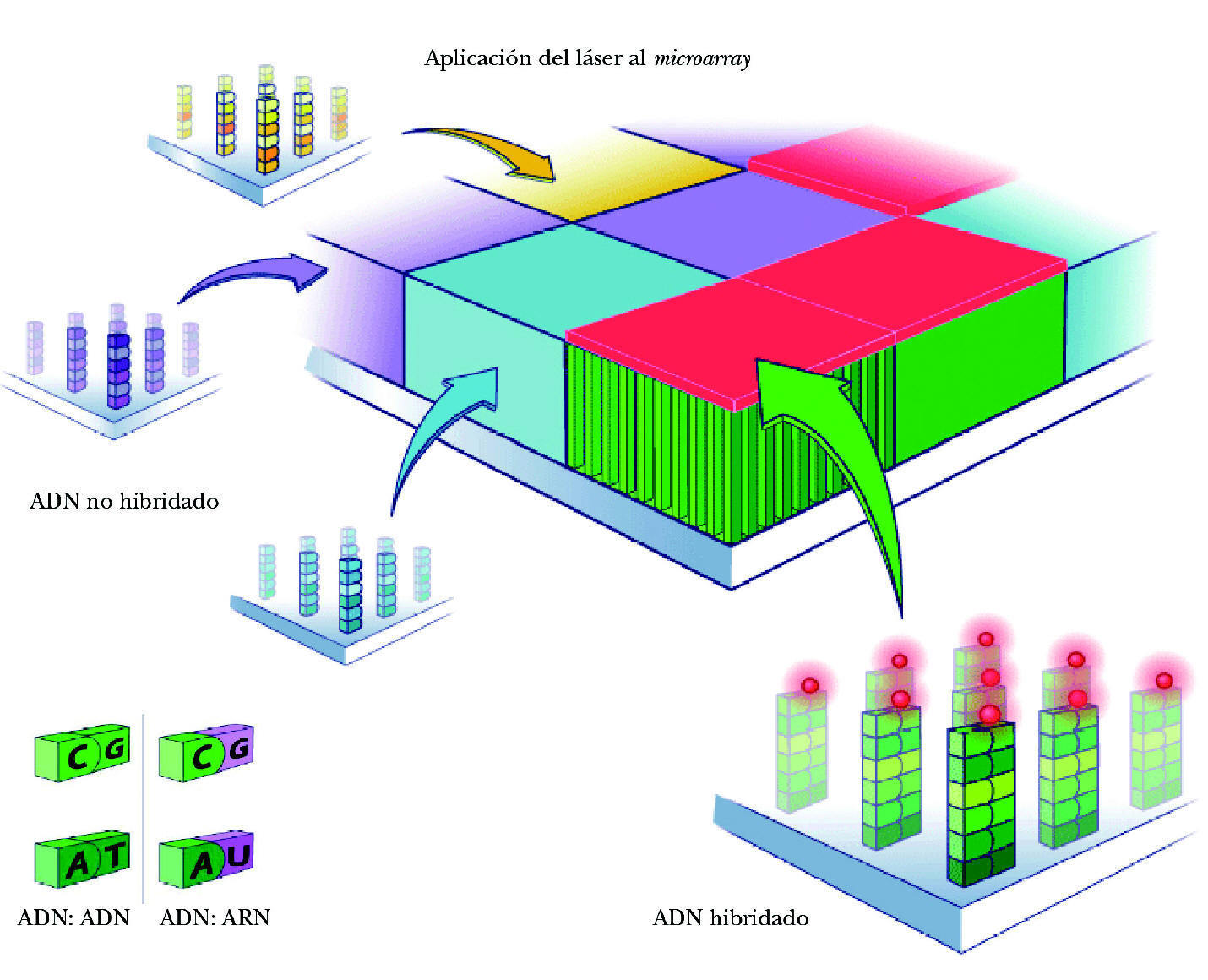
Figura 6. Hibridación del ADN.
También se utilizan moléculas fluorescentes que se unirán a la biotina, combinada, asimismo, a las sondas de ADN. La lectura de la fluorescencia y sus diferentes intensidades también se realiza con un láser escáner completando mapas de calor. Con ello se identificarán los genes y los polimorfismos que posea el paciente a través de sus muestras. Valorando todas las cuadrículas se podrán evaluar todos los varios miles de genes presentes en el array. Conociendo la identidad de las sondas de oligonucleótidos (y la de sus genes correspondientes) por su localización en el microarray podremos identificar los genes y sus polimorfismos presentes. Basándose en el patrón de hibridación también se podrá identificar si un SNP es homo o heterocigótico.
Objetivos del estudio de los polimorfismos
El estudio de los polimorfismos tiene como objetivos:
1.Determinar marcadores genéticos de susceptibilidad a una enfermedad. El análisis de polimorfismos permite determinar la predisposición de un individuo a padecer una enfermedad. Se pueden encontrar el gen o grupo de genes «candidatos» que contribuyen a padecer la enfermedad.
2.Determinar la predisposición de un individuo a un determinado subtipo dentro de una enfermedad. Para enfermedades con clínica muy heterogénea, como la psoriasis, donde la carga genética puede ser parcialmente responsable de tal variabilidad clínica.
3.Estudiar el desarrollo de tumores y su progresión. Se ha podido comprobar cómo los polimorfismos tipo SNP intervienen en la biología tumoral.
4.Identificar dianas terapéuticas. Al igual que sucede con los análisis de expresión génica, el estudio del genotipo también contribuye a la farmacogenómica.
5.Predecir la respuesta terapéutica a un fármaco. La variabilidad en la respuesta farmacológica genéticamente determinada tiene mucho que ver con polimorfismos en genes específicos. Según diferentes estudios, algunos SNP se han relacionado con cambios en el metabolismo, proteínas transportadoras y receptores de fármacos, grado de respuesta de algunos fármacos y porcentaje de toxicidad. De hecho, algunos SNP ya se están utilizando para predecir la respuesta clínica a los fármacos 11-14. Los individuos portadores de un determinado alelo probablemente lo son también de variantes específicas con varios marcadores de SNP. Por ello, para predecir la respuesta a un medicamento no será necesario identificar los verdaderos genes y alelos implicados, sino que sería suficiente con detectar los SNP.
En un futuro los avances tecnológicos asociados con la robótica y las reacciones múltiples reducirán significativamente el coste del análisis de varios cientos de miles de SNP por paciente en ensayos clínicos. Los patrones bioinformáticos permitirán la rápida comparación de estos patrones de SNP entre los pacientes que presenten diferencias fenotípicas en la respuesta a un fármaco, de tal manera que cuando la información de las diferencias en la respuesta farmacológica sea ostensible, los perfiles de los SNP podrán ser relacionados con los actuales chips diagnósticos, y así determinar o anticipar la respuesta a un fármaco en un paciente determinado.
Resecuenciación génica
Los análisis de resecuenciación génica permiten identificar el ADN de la muestra de un organismo o incluso de un virus. Es una herramienta de identificación (por ejemplo al determinar la cepa de virus en una epidemia, localizar agentes patógenos en muestras de agua o alimentos, determinar si un alimento contiene sólo ese alimento y no ha sido mezclado con otros, etc.). Son análisis bastante complicados y que se escapan del objetivo de esta revisión.
Farmacogenética y bioinformática
La bioinformática es la «organización, manipulación y análisis de resultados en biología molecular y biomedicina utilizando métodos computacionales», es decir, pone orden cognitivo en la información acumulada. Aunque su origen hay que situarlo hace 30 años, es evidente que su importancia y el papel central que ahora desempeña en la investigación en biomedicina está directamente relacionado con la explosión de las técnicas de genómica. Su desarrollo se ha realizado al hilo de las necesidades generadas por la rápida evolución de las técnicas experimentales, particularmente en el caso de la genética molecular, donde la información sobre la secuencia de genes y proteínas ha requerido desde el principio la organización, almacenamiento y análisis de la información generada experimentalmente 15.
En una primera aproximación, el problema al que se enfrenta la bioinformática se relaciona con el análisis de la secuencia de ácidos nucleicos que comparten los genomas. Contamos en la actualidad con 31 millones de entradas en la base de datos, 45.000 millones de bases y un millón de entradas de proteínas con 310 millones de aminoácidos. Esta cantidad de datos crea problemas computacionales. Pero es que además, la biología molecular y la biomedicina generan no sólo muchos datos, sino datos muy diversos, lo que plantea problemas bastante difíciles durante la generación de las correspondientes estructuras de bases de datos (métodos de almacenamiento) que deben adaptarse constantemente a los nuevos métodos.
Los dos principales centros que mantienen las bases de datos esenciales son el NCBI en EE.UU. y el EBI-EMBL en Europa, aunque muchos otros producen bases de datos focalizadas en diversos temas. Las principales bases de datos en biología molecular y biomedicina incluyen:
1.National Center for Biotechnology Information (NCBI). Parte de la National Library for Medicine del NIH (www.ncbi.nlm.hih.gov).
2.European Bioinformatics Institute (EBI-MBL) (www. ebi.ac.uk).
3.Sanger Center (www.sanger.ac.uk).
4.Grupo de Diseño de Proteínas (PDG, CNB-CSIC) (www.pdg.cnb.uam.es).
5.Secuencias de ADN (www.ebi.ac.uk.embl).
6.Secuencias de proteínas (www.ebi.ac.uk.embl).
7.Dominios de proteínas (www.ebi.ac.uk/interpro/index.html).
8.Estructuras tridimensionales (www.ebi.ac.uk/MSD).
9.Bases moleculares de enfermedades (www.ncbi.nlm. hih.gov/omim).
10.Un sistema para navegar entre bases de datos (http:// srs.ebi.ac.uk).
Por otra parte, varios sistemas públicos y privados contienen métodos para el análisis de los resultados de los microarrays de ADN, incluyendo la creación de estándares para la comparación de resultados generados por distintos laboratorios.
En el caso concreto de la farmacogenética, la aportación de la bioinformática incluye: a) organización y almacenamiento de la información experimental generada; b) diseño de bases de datos adaptadas a la tecnología empleada en farmacogenética; c) capacidad de almacenar tanto imágenes como datos obtenidos en los estudios realizados; d) estimación de la significación estadística de los resultados; e) comparación de los patrones de expresión entre varias muestras (del mismo paciente o entre pacientes), y f) análisis de resultados incluyendo la comparación de éstos con la información sobre funciones, enfermedades y medicamentos disponible, para predecir con una determinada fiabilidad estadística la efectividad de un medicamento y su tolerancia en un paciente determinado.
Farmacogenética y aspectos éticos
La utilización de pruebas genéticas para diagnosticar o predecir enfermedades, para predecir la respuesta a tratamientos o valorar los posibles efectos adversos a los mismos, constituirá en un futuro no muy lejano una herramienta médica para mejorar la salud de la población a nivel individual, a partir del conocimiento de la susceptibilidad genética para padecer ciertas enfermedades o la respuesta terapéutica. Ahora bien, la información que podría obtenerse como consecuencia de la realización de estas pruebas genéticas plantea problemas relativos a esa misma información, a su acceso y a su utilización, pues pueden entrar en conflicto los intereses del individuo afectado con los de otras personas o entidades. Las inquietudes se centran en decidir quién, bajo qué circunstancias y con qué objetivos se podrán realizar pruebas genéticas tras la obtención de muestras biológicas para su realización; quién tendrá acceso a la información obtenida, a quién podrá comunicarse y en qué circunstancias, qué utilización se podrá dar a la misma y qué medidas de protección a este respecto deberían adoptarse 16. Todo ello abre un debate ético en el que, si bien se ven especialmente involucrados el diagnóstico y predicción del riesgo a padecer enfermedades (relación con los seguros de enfermedad, puestos de trabajo, etc.), también incumbe a la farmacogenómica y la farmacogenética.
Por ello, es necesaria una regulación legal que aborde estas cuestiones. El Estado español ha dado ya pasos en este sentido a través de la ratificación del Convenio del Consejo de Europa para la protección de los derechos humanos y la dignidad del ser humano con respecto a las aplicaciones de la biología y la medicina, llevado a cabo en Oviedo el 4 de abril de 1997 (BOE de 20 de octubre de 1999), y conocido como Convenio de Oviedo, que establece de forma clara que el interés y el bien del ser humano deben prevalecer sobre el solo interés de la sociedad y la ciencia. Dada la necesidad de respeto a la prevalencia de los intereses individuales y el derecho a la libertad, el proceso de realización de una prueba genética debe ajustarse a las exigencias contempladas en la Ley General de Sanidad, de 1986, entre las que se encuentra el consentimiento informado, que debe incluir información relativa a la naturaleza de los análisis, los objetivos perseguidos con la prueba y las consecuencias y posibilidades de actuación que se pueden aplicar en caso de un resultado positivo. Actualmente con el consentimiento se establece que el control de los datos obtenidos corresponde al titular. El individuo tiene derecho a decidir por sí mismo sobre la utilización de los datos con la potestad de poder acceder a los mismos, al tiempo que controla su existencia y veracidad, y autorizar su revelación. Esto consagra la prevalencia del derecho a la intimidad genética sobre cualquier otro derecho. Ninguna persona física, ni entidades públicas o privadas, deberán tener derecho a analizar la información genética de una persona sin su consentimiento. El propietario de la información genética es la persona física. Por tanto, cualquier decisión relativa a la difusión de dicha información a terceros, incluyendo los familiares genéticos, debe ser estrictamente personal 17.
La Ley Orgánica 15/1999 de Protección de Datos de Carácter Personal amplía la protección a toda clase de datos de esta índole, y no sólo a los informatizados, lo que permite su aplicación a cualquier material de origen biológico que contenga información personal identificable, y legitima la existencia de bases de datos genéticas con registros informáticos. Esto conlleva que si se establecen registros genéticos, el almacenamiento de datos debería ser voluntario y se debería garantizar la estricta confidencialidad de los mismos. Los responsables del sistema deben garantizar la imposibilidad de acceder a los datos genéticos y personales, su destrucción o difusión sin el consentimiento expreso de la persona titular de ellos 18.
También, tanto el Convenio del Consejo de Europa (4 de abril de 1997) sobre Derechos Humanos y Biomedicina (artículos 5, 10.1 y 12) como la Declaración Universal sobre el Genoma Humano y los Derechos Humanos (11 de noviembre de 1997) de la UNESCO (artículo 5.b), establecen que en todos los casos se recabará el consentimiento previo, libre e informado de la persona interesada, como derecho fundamental del individuo 19. En este contexto, la UNESCO promulgó en octubre de 2003 una declaración internacional sobre protección de datos genéticos humanos, con el ánimo de evitar el uso discriminatorio de los genes que predisponen a diversos tipos de enfermedades. Podría hacerse extensivo a su relación con la respuesta terapéutica. En función de estas recomendaciones, en la inmensa mayoría de países europeos se están poniendo en práctica en los últimos años una serie de normativas y recomendaciones para el empleo correcto de las pruebas genéticas.
Por último, conviene tener presente que los planteamientos médicos, éticos y legales en relación con la utilización de pruebas genéticas deberán ser revisados y corregidos periódicamente a medida que avanza la ciencia y evoluciona la sociedad. La comunidad científica deberá desempeñar un papel clave en concienciar a los pacientes y, por tanto, a la opinión pública acerca del correcto uso de estas nuevas tecnologías.
Conflicto de interesesDeclaramos no tener ningún conflicto de intereses
Correspondencia:
Esteban Daudén Tello.
Servicio de Dermatología.Hospital Universitario de La Princesa.
Diego de León, 62. 28006 Madrid.
Aceptado el 23 de octubre de 2006.


