









INTRODUCCIÓN
Los linfomas cutáneos primarios de células B (LCCB) representan el 20-25 % del total de los linfomas cutáneos. Constituyen un grupo heterogéneo de entidades, por lo general con un comportamiento clínico indolente, aunque las recurrencias cutáneas no son excepcionales. Sólo ocasionalmente puede observarse el desarrollo de enfermedad extracutánea1,2 .
Se desconocen los mecanismos implicados en la presencia y la persistencia de estas células B neoplásicas malignas en la dermis. Sólo en algunos casos se han relacionado con una estimulación antigénica persistente debido a la presencia de algunos agentes infecciosos como virus (herpes humano tipo 8 [VHH-8], de la inmunodeficiencia humana [VIH], de la hepatitis C [VHC]) o bacterias (Borrelia burgdorferi) o en el contexto de enfermedades autoinmunes 3,4 .
Hasta el momento actual no se han identificado lesiones moleculares específicas en las formas cutáneas primarias y tampoco se ha demostrado la presencia de las alteraciones genéticas conocidas e identificadas como específicas de linfomas no hodgkinianos ganglionares o extraganglionares. Por ejemplo, la translocación t(14;18)(q32;q21), detectada entre el 70 y el 90 % de los linfomas foliculares ganglionares, sólo se ha demostrado excepcionalmente en los linfomas foliculares primarios cutáneos. Tampoco existe ninguna evidencia de la participación de la translocación t(11;18)(q21;q21), característica de los linfomas MALT (tejido linfoide asociado a mucosas) extraganglionares, en los linfomas cutáneos de la zona marginal 5 .
CLASIFICACIÓN
Durante los últimos 15 años se han propuesto y utilizado diversos criterios diagnósticos para designar los distintos subtipos de LCCB. Las clasificaciones actuales de las neoplasias hematológicas intentan individualizar entidades clinicopatológicas basándose en sus características clínicas, histopatológicas, inmunofenotípicas y genotípicas. Tal como se comentó en el artículo dedicado a los linfomas cutáneos de células T, la reciente clasificación de la Organización Mundial de la Salud (OMS) de los linfomas es probablemente la más actual y exhaustiva 6-8 . Esta clasificación reconoce también las peculiaridades que tienen los linfomas que se originan primariamente en la piel. El consenso y uniformidad de criterios alcanzado en esta nueva clasificación permiten una mejor metodología en la elaboración de estudios y comunicación de resultados. Sin embargo, desde el punto de vista dermatológico, la clasificación más utilizada en la actualidad es la clasificación de la European Organization for Research and Treatment of Cancer (EORTC), una clasificación específica de los procesos linfoproliferativos cutáneos primarios, que es la que se sigue en este artículo (tabla 1) 9-11 .

TABLA 1. CLASIFICACIÓN DE LOS LINFOMAS CUTÁNEOS PRIMARIOS DE CÉLULAS B
La clasificación de la EORTC establece dos categorías en los LCCB de curso indolente: a) de la zona marginal (LCBZM), y b) del centro folicular o linfoma folicular (LCBCF) (tabla 1). La similitud clínica e histológica de los LCBZM con los linfomas tipo MALT ha motivado que algunos autores propusieran el término de linfomas SALT (tejido linfoide asociado a piel) para designar este grupo de procesos. El grupo de LCCB de agresividad intermedia estaría formado por los linfomas cutáneos primarios de células B localizados en las piernas (LCBP) y, por último, se consideran unas entidades de carácter provisional por su escasa casuística, el linfoma B intravascular y el plasmacitoma cutáneo 9,12,13 .
METODOLOGÍA DIAGNÓSTICA
Aspectos clínicosLos LCCB primarios parecen ser más frecuentes en mujeres, en una proporción 2:1, y suelen manifestarse aproximadamente a los 60 años. Con independencia del subtipo, en la mayoría de los pacientes con un LCCB primario, las lesiones cutáneas corresponden clínicamente a pápulas o nódulos eritematosos, violáceos o del color de la piel normal, y discretamente infiltrados al tacto. En ocasiones se observan placas infiltradas
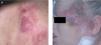
o lesiones arciformes (anulares) rodeadas por nódulos en la periferia. Dichas lesiones suelen localizarse en el tronco, ocasionalmente en la región cefálica, y con menos frecuencia en las extremidades. Existe una cierta predilección regional para algunos subtipos específicos de LCCB: los linfomas foliculares se localizan con mayor frecuencia en el cuero cabelludo, las lesiones del LCBZM suelen desarrollarse en el tronco y las extremidades y el linfoma B de células grandes de curso agresivo suele localizarse en las piernas (fig. 1) 9,14,15 .Aspectos histopatológicos y de diagnóstico molecular
El diagnóstico de un LCCB primario y la diferenciación respecto a otras enfermedades que se asemejan a procesos linfoproliferativos cutáneos de células B (pseudolinfomas B o hiperplasias linfoides reactivas) puede resultar difícil si únicamente se basa en los hallazgos histopatológicos. Sin embargo, cada subtipo de LCCB presenta unos hallazgos morfológicos característicos que, valorados en combinación con criterios inmunofenotípicos y genotípicos, suelen permitir establecer el diagnóstico definitivo.
Desde un punto de vista histológico, las lesiones precoces muestran un infiltrado linfoide nodular o de características parcheadas en distribución perivascular y perianexial en la dermis superficial. En las lesiones más avanzadas suele observarse un infiltrado más denso y difuso que se extiende desde la dermis al tejido celular subcutáneo, con folículos linfoides reactivos o sin ellos. Puede existir una infiltración masiva y una destrucción de las estructuras anexiales (lesión linfoepitelial). La epidermis se halla característicamente preservada y suele existir una zona de colágeno normal (zona grenz) que separa el infiltrado linfoide de la epidermis normal.
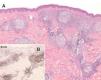
El estudio inmunofenotípico suele poner de manifiesto que las células neoplásicas expresan antígenos linfoides de células B (CD19, CD20, CD22 y CD79a). En ocasiones se detecta un inmunofenotipo B aberrante, caracterizado por la coexpresión de CD43 y CD20. En los linfomas B cutáneos un indicador de clonalidad que se descubre con frecuencia mediante técnicas inmunohistoquímicas es la presencia de una expresión monotípica de cadenas ligeras de las inmunoglobulinas, kappa () o lambda (). En los procesos reactivos la proporción habitual : es 2:1. Una ratio superior a 5-10:1 (restricción ) o una proporción inferior a 0,5-1:1 (restricción ) indican clonalidad (fig. 2) 1,16 .
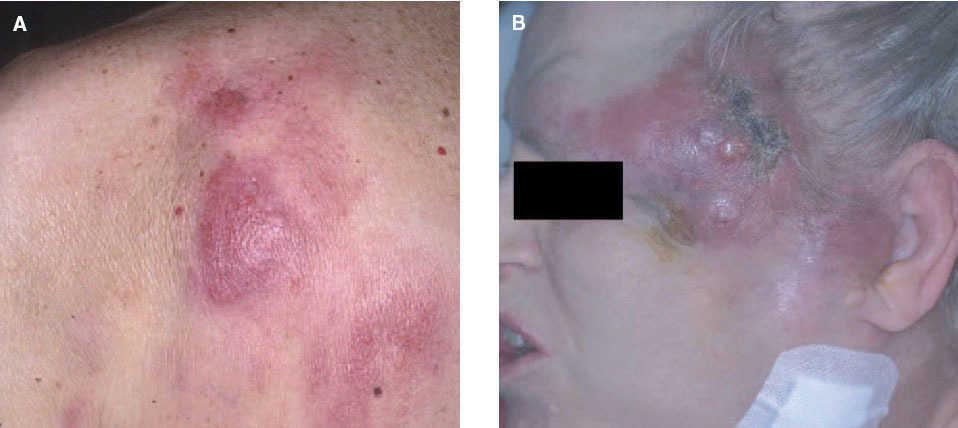
Fig. 1. A) Linfoma cutáneo B folicular secundario. B) Linfoma cutáneo B difuso secundario.
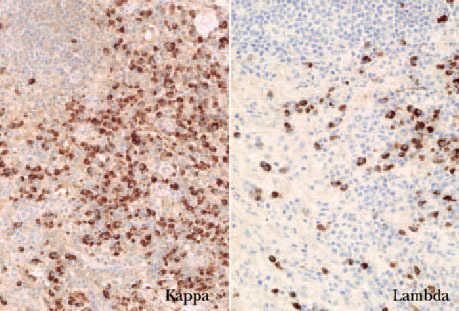
Fig. 2.Expresión monotípica de cadenas ligeras (/, ×100).
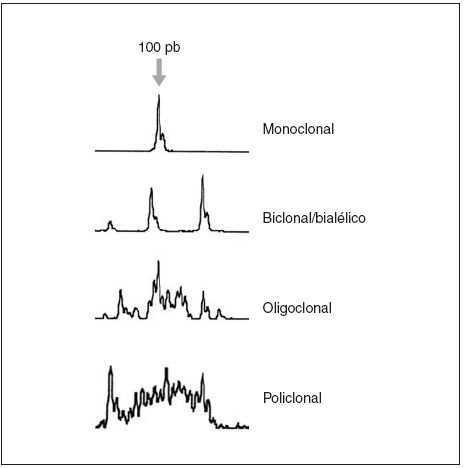
Fig. 3.Patrones de reordenamiento del gen de las cadenas pesadas de las inmunoglobulinas y análisis mediante Genescan.
La demostración de un reordenamiento monoclonal del gen de las cadenas ligeras o pesadas de las inmunoglobulinas mediante la técnica de Southern blot y/o por reacción en cadena de la polimerasa (PCR) apoya el diagnóstico de linfoma B (fig. 3).
Estudio de extensiónUna vez establecido el diagnóstico, la valoración de un paciente con un LCCB debe incluir una historia clínica y una exploración física detallada con el objetivo de descartar una posible afectación sistémica (presencia de síntomas y signos B, palpación minuciosa de los ganglios linfáticos, o descartar la presencia de visceromegalias). El estudio de extensión debe incluir recuento y fórmula de sangre periférica; perfil bioquímico convencional (incluyendo los niveles de lacticodeshidrogenasa [LDH] y 2-microglobulina); radiografía de tórax; tomografía computarizada (TC) toracoabdominal, y biopsia de médula ósea en todos los casos (tabla 2).

TABLA 2. CONDUCTA QUE DEBE SEGUIRSE EN EL DIAGNÓSTICO ESTADIFICACIÓN DE LOS LINFOMAS CUTÁNEOS DE CÉLULAS B
Diagnóstico diferencial
El diagnóstico diferencial siempre debe establecerse con los pseudolinfomas B o hiperplasias linfoides B reactivas. Esta diferenciación puede resultar difícil, sobre todo en los linfomas de células pequeñas y con una intensa formación de centros germinales como los LCBCF y los LCBZM. En estos casos es necesario emplear técnicas inmunohistoquímicas y de biología molecular (estudio de clonalidad) para poder establecer el diagnóstico definitivo. En los pseudolinfomas se observa una expresión politípica de las cadenas ligeras de las inmunoglobulinas, así como un reordenamiento policlonal linfoide B 17 .
SUBTIPOS INDOLENTES
Linfoma cutáneo primario de células B de la zona marginal/inmunocitoma primario cutáneoEl concepto de LCBZM define un subtipo de linfomas formados por linfocitos de pequeño a mediano tamaño con morfología variada, de tipo centro-cito (pequeñas de núcleo hendido), con células B monocitoides (pequeñas de citoplasma amplio acuoso) y con cierto grado de diferenciación plasmacitoide (células plasmáticas y células linfoplasmocitarias). El término de inmunocitoma define de manera exclusiva un subgrupo morfológico de LCBZM en el que predomina la diferenciación plasmacitoide.
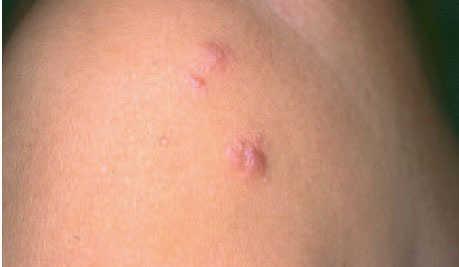
Fig. 4.Lesiones nodulares correspondientes a un linfoma cutáneo primario de células B de la zona marginal.
Los linfomas de la zona marginal (tipo MALT, MAL-Toma o SALToma) se han descrito con frecuencia en localizaciones extraganglionares o en mucosas como el estómago, las glándulas salivales, el tiroides, la mama, el pulmón y la tráquea, el cuello uterino, así como en la piel y en el tejido celular subcutáneo. Probablemente el LCBZM representa la forma más frecuente de LCCB primario2,18,19 .
Diagnóstico
Aspectos clínicos. Se presentan clínicamente como nódulos o placas eritematosas solitarias o múltiples, a menudo localizadas en el tronco o en la parte proximal de las extremidades. Afecta a pacientes de edad mediana y no muestra una clara predisposición por ningún sexo. Su comportamiento es indolente permaneciendo localizados en la piel y raramente desarrollan diseminación extracutánea. La evolución clínica suele ser favorable, con una baja tasa de mortalidad (supervivencia superior al 95 % a los 5 años), aunque son relativamente frecuentes las recurrencias cutáneas. Su comportamiento clínico es bastante similar al de otros linfomas de la zona marginal de localización extracutánea (fig. 4)2,20-24 .
Aspectos histopatológicos. Los LCBZM se caracterizan citológicamente por una proliferación neoplásica linfoide, siguiendo un patrón de infiltración nodular o difuso que suele adoptar una distribución perianexial en la dermis reticular. En el tejido celular subcutáneo forma largos cordones tumorales. Habitualmente respeta la epidermis y relativamente la dermis papilar. Se observan centros germinales reactivos bien formados o rudimentarios, y en ocasiones un fenómeno de colonización folicular por parte de los linfocitos neoplásicos. La infiltración del epitelio glandular, o lesión linfoepitelial, es poco frecuente en los LCBZM. Este fenómeno es un hallazgo característico de los LCBZM extraganglionares. El infiltrado neoplásico posee una composición celular heterogénea y variable: linfocitos de tamaño pequeño o intermedio con un citoplasma pálido moderadamente abundante (células B monocitoides), células similares a centrocitos, blastos transformados de gran tamaño (similares a centroblastos) y proporciones variables de células con diferenciación plasmacitoide. Las células plasmáticas presentan inclusiones intranucleares, denominadas cuerpos de Dutcher, patognomónicas de neoplasias de estirpe B, fácilmente visibles con una tinción de ácido peryódico de Schiff (PAS). Existe un fenómeno de compartimentalización característico de las células plasmáticas en la región subepidérmica, en la periferia de los agregados linfoides y en las zonas interfoliculares24,25.
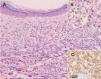
Características inmunohistoquímicas. Las células neoplásicas de los linfomas de la zona marginal no presentan un marcador inmunofenotípico característico o diagnóstico. Estas células corresponden a linfocitos B (CD20+, CD22+, CD43+/–, CD11c+/–), que no expresan ni los antígenos CD5, ciclina-D1 (marcadores de los linfocitos pequeños del manto) ni el antígeno CD10 (característico de los linfomas centrofoliculares) y habitualmente tampoco expresan el antígeno CD23 (característico de las células de la leucemia linfática crónica). Las células tumorales pueden expresar KiM1p (marcador monocitoide), IgM y, ocasionalmente, IgA e IgG. Las células plasmáticas y linfoplasmacitoides expresan el antígeno CD79a. Estas células plasmáticas pueden mostrar una expresión monotípica de las cadenas ligeras de las inmunoglobulinas. Las células de la zona marginal y plasmáticas monotípicas (neoplásicas) suelen localizarse en las regiones interfoliculares (parafoliculares), siendo los folículos de origen reactivo y no neoplásico (lo que permite diferenciarlos de los linfomas cutáneos B centrofoliculares), aunque ocasionalmente puede existir una invasión o colonización neoplásica de los folículos reactivos que dificultan en gran medida el diagnóstico específico (fig. 5)2,25.
Aspectos genéticos y moleculares. Los LCBZM representan proliferaciones linfoides clonales. La detección del reordenamiento del gen de las cadenas pesadas de las inmunoglobulinas (IgH) mediante la técnica de PCR demuestra una población clonal linfoide B en el 50-70 % de los casos de LCBZM2,26 .
No existen alteraciones genéticas conocidas específicas en los LCBZM. Sólo en un porcentaje limitado de casos se han comunicado algunas alteraciones genéticas. Recientemente se ha descrito la translocación 11;18(q21,q21) que fusiona el gen inhibidor de la apoptosis API2 con el gen MLT (MALT-1) , en algunos subtipos de linfomas de la zona marginal (linfomas MALT gástricos y pulmonares). Por otro lado, excepcionalmente se han descrito mutaciones puntuales del gen BCL10 (con una cierta actividad proapoptótica),
o la síntesis de una proteína BCL-10 truncada como consecuencia de la t(1;14)(p22;q32). Sin embargo, la positividad nuclear (expresión aberrante) para la proteína BCL-10 parece relacionarse con la t(11;18) (q21;q21), por lo que se ha sugerido que tanto el gen BCL10 como los transcritos API2-MALT1 podrían interactuar en la patogénesis de los linfomas MALT. Finalmente, se ha descrito recientemente que los linfomas MALT extraganglionares que presentan la translocación t(11;18)(q21;q21), 30-50 % de los casos, pueden evolucionar a linfomas difusos de células grandes y cuando son de origen gástrico parecen relacionarse con la resistencia al tratamiento erradicador de Helicobacter pylori , requiriendo con frecuencia tratamiento quimioterápico. En los pocos trabajos existentes en LCBZM no se ha demostrado la participación de esta alteración citogenética5,27-29 .
Los LCBZM pueden expresar de forma variable la proteína BCL-2, sin que parezca que conlleve implicaciones pronósticas. Tampoco presentan la translocación t(14;18) que implica a los genes BCL2/IgH , característica de los linfomas foliculares ganglionares. Recientemente se ha descrito la t(14;18)(q32;q21) que yuxtapone el gen IgH con el gen MALT-1 , con una cierta especificidad para linfomas MALT de localización extraganglionar30 .
Valoración
La observación de lesiones con unas características clínicas e histopatológicas compatibles con LCBZM obliga a descartar una afectación secundaria cutánea a partir de un linfoma ganglionar o extraganglionar, similar a todos los LCCB (tabla 2). Ocasionalmente puede solicitarse un estudio serológico que incluya determinaciones de virus linfotropos (virus de Epstein-Barr, citomegalovirus, VIH, VHH-8), VHC y de anticuerpos frente a B. burgdorferi . De forma excepcional, los LCBZM se asocian a enfermedades autoinmunes4 .
Diagnóstico/diagnóstico diferencial
El diagnóstico de LCBZM plantea importantes dificultades, sobre todo en su diferenciación con procesos reactivos (hiperplasias linfoides reactivas) con los que puede compartir unas características histopatológicas e inmunohistoquímicas comunes. De forma invariable suele existir un infiltrado linfoide T reactivo entremezclado con las células neoplásicas que en algunas situaciones puede ser muy denso o predominante (LCCB rico en células T). Un infiltrado denso linfocítico de células pequeñas, con histiocitos y células inflamatorias agudas y escasas células plasmáticas, requiere descartar la presencia de agentes infecciosos o fármacos. La presencia de eosinófilos y vénulas en el tejido interfolicular puede confundirse erróneamente con la enfermedad de Kimura, una hiperplasia angiolinfoide subcutánea o con una picadura persistente de artrópodo 31 .
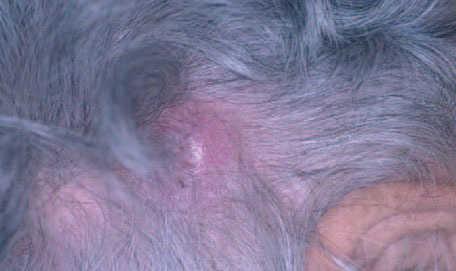
Fig. 6.Nódulo en cuero cabelludo correspondiente a un linfoma cutáneo primario B folicular.
Sólo la combinación de los hallazgos clinicopatológicos, las características inmunofenotípicas (restricción de cadenas ligeras de las inmunoglobulinas) y/o la demostración de clonalidad (análisis genotípico), junto con la evolución clínica permite establecer un diagnóstico definitivo.
Tratamiento
La aproximación terapéutica de los LCBZM no suele plantear dificultades. En las lesiones cutáneas localizadas o recurrentes, la exéresis quirúrgica y/o la radioterapia localizada suelen ser el tratamiento de elección y, de forma menos habitual, la administración intralesional de quimioterápicos (cisplatino). En casos con una enfermedad cutánea extensa podría plantearse el uso de monoterapia con rituximab (anticuerpo monoclonal anti-CD20) o diversas pautas de poliquimioterapia. El curso clínico es favorable, aunque ocasionalmente desarrollan recurrencias cutáneas 9,32,33 .
Linfomas primarios cutáneos de células del centro folicular (linfomas foliculares)
Se consideran linfomas foliculares cutáneos aquellos linfomas de células B que tienen un patrón de crecimiento folicular y/o están compuestos por células del centro folicular (grandes centrocitos y centroblastos). A este respecto, uno de los aspectos más criticable de la clasificación de la EORTC es quizás una definición poco rigurosa de éstos. Si bien desde un punto de vista morfológico los linfomas foliculares tienen un patrón de crecimiento folicular, los de origen cutáneo suelen adoptar uno de infiltración difuso. En aquellos casos en los que se observan folículos, suelen estar formados por verdaderas células neoplásicas de origen centro folicular. La expresión de BCL-2 por parte de las células neoplásicas es poco frecuente, así como la presencia de la translocación t(14;18), característica de los linfomas foliculares ganglionares. En nuestro medio no es habitual encontrar esta alteración citogenética y su presencia obliga a descartar la presencia de un linfoma sistémico subyacente34,35 .
Diagnóstico
Aspectos clínicos. Los LCBCF se presentan clínica-mente en forma de placas o nódulos solitarios o múltiples que a menudo afectan a la región de la cabeza y el cuello, por lo general en personas de edad avanzada. Suelen limitarse a la piel durante largos periodos de tiempo, presentan un comportamiento benigno y pueden aplicarse en ellos tratamientos locales de acción directa sobre la piel (fig. 6).
Aspectos histológicos y genéticos. Los LCBCF muestran un infiltrado nodular o difuso sin epidermotropismo y con un aspecto histológico variable. Las células neoplásicas corresponden a células centrocíticas grandes (con núcleo denso y hendido) y centroblastos e inmunoblastos (de núcleo grande no hendido y nucléolo prominente). La relativa proporción de cada una de estas células depende del grado de proliferación, tamaño y tiempo de evolución de las lesiones, sin que haya correlación significativa con el pronóstico. Es típico encontrar la combinación de zonas con características histopatológicas de bajo y alto grado en lesiones de un mismo paciente o incluso en diferentes áreas de una misma lesión36 .
Las células neoplásicas expresan los antígenos CD19, CD20, CD22 y a menudo muestran restricción de cadenas ligeras de las inmunoglobulinas. La expresión de la proteína BCL-2 suele ser negativa, y tampoco expresan los antígenos CD5, CD11c, ni CD43. Un hallazgo característico es la positividad para antígenos de células centrofoliculares como el CD10 o el BCL-6, aunque este fenómeno ha sido recogido de forma variable e inconstante en la literatura especializada (fig. 7)37 .
Con frecuencia se demuestra el reordenamiento monoclonal del gen de la IgH mediante PCR. La demostración de mutaciones somáticas en la región VH del gen de la IgH o del gen BCL6 corroboran el origen centrofolicular de estos linfomas38,39 . La translocación t(14;18) es poco frecuente y tampoco hay estudios que impliquen otras alteraciones genéticas específicas descritas en los linfomas foliculares sistémicos (translocaciones del gen BCL6 o alteraciones citogenéticas estructurales y numéricas en diversos cromosomas, como por ejemplo +7, +18, 3q27-28, 6q23-26, 17p, 9p)37,40,41 .
Diagnóstico diferencial
La observación de un LCCB con patrón de crecimiento folicular con células neoplásicas centrofoliculares (CD10+/BCL-6+/BCL-2+) obliga a descartar la presencia de un linfoma cutáneo B folicular secundario, en particular si se demuestra la presencia de la t(14;18).
Su diferenciación con los LCBZM puede ser controvertida. Muchos autores han propuesto un origen celular en la zona marginal (parafolicular/monocitoi-de) para la mayor parte de los LCCB, lo cual implicaría que los LCBCF deben expresar en todos los casos los antígenos CD10 y/o BCL-6.
El diagnóstico diferencial de un linfoma cutáneo con patrón de crecimiento folicular debe también incluir las hiperplasias linfoides reactivas. La demostración de un fenotipo aberrante y de una población monoclonal linfoide B permitiría establecer su carácter neoplásico, mientras que una población celular heterogénea iría más a favor de un proceso reactivo42 .
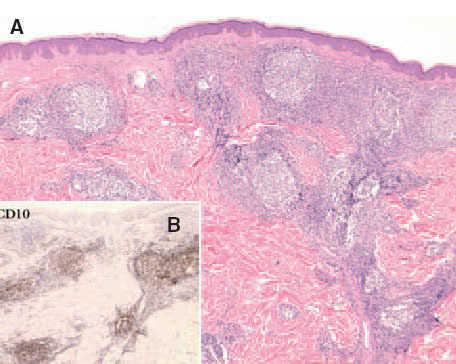
Fig. 7. A) Infiltrado linfoide dérmico con patrón de crecimiento folicular de un linfoma folicular secundario. (Hematoxilina-eosi-na, ×100.) B) Detalle de la expresión del antígeno de células del centro folicular CD10 (CD10, ×100).
Aspectos pronósticos y terapéuticos
La mayoría de los LCBCF suelen ser neoplasias linfoides de comportamiento benigno, aunque presentan un patrón infiltrativo difuso. El patrón de infiltración no necesariamente se correlaciona con el pronóstico. Los LCBCF difusos de localización craneofacial o truncal suelen seguir una evolución indolente, aunque los que se localizan en las extremidades inferiores parecen tener un pronóstico menos favorable.
La mayoría de los pacientes pueden ser tratados mediante cirugía o radioterapia local, siendo el pronóstico excelente con una supervivencia a los 5 años próxima al 100 %. Al igual que en el caso de los LCBZM existe la posibilidad de tratamiento mediante rituximab con una respuesta clínica variable35,37 .
LINFOMAS CUTÁNEOS DE CÉLULAS B DE AGRESIVIDAD INTERMEDIA
Linfoma primario cutáneo B de célula grande localizado en las piernasEl linfoma primario cutáneo B difuso de célula grande que aparece en las extremidades inferiores se caracteriza por un pronóstico poco favorable con un número elevado de recaídas y una mayor afectación extracutánea. Afecta principalmente a pacientes de edad avanzada y presenta cierto predominio por las mujeres. Para algunos autores, la localización anatómica no se considera un factor fundamental y se ha especulado que otras variables (predominio de la morfología celular redonda sobre la hendida, presencia de múltiples lesiones cutáneas, expresión de la proteína BCL-2) podrían tener mayor valor pronóstico (fig. 8)43 .
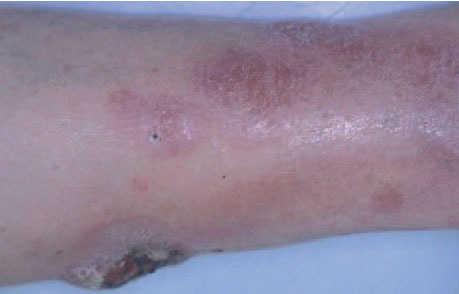
Fig. 8.Placa infiltrada con desarrollo de lesión tumoral ulcerada en un paciente con un linfoma B de célula grande de las piernas.
Histológicamente se observa un predominio de centroblastos e inmunoblastos. Aquellos casos que presentan una población celular homogénea de inmunoblastos (nucléolo prominente) se pueden sub-clasificar como linfomas B inmunoblásticos (fig. 9). Las células expresan habitualmente los antígenos CD19, CD20, CD22 y CD79a (antígenos pan-B). La expresión de CD5 y CD10 no es constante, y ocasionalmente pueden expresar inmunoglobulinas citoplasmáticas o de superficie. Es frecuente observar una expresión marcada de la proteína BCL-2. Puede detectarse reordenamiento clonal del gen de la IgH. Sin embargo, la expresión de BCL-2 no se relaciona con la presencia de la t(14;18)44 .
Presenta una supervivencia del 60 % a los 5 años. Este dato comporta que pueda plantearse un tratamiento con poliquimioterapia, aunque las lesiones solitarias o localizadas podrían ser tratadas inicialmente de forma más conservadora (radioterapia)45 .
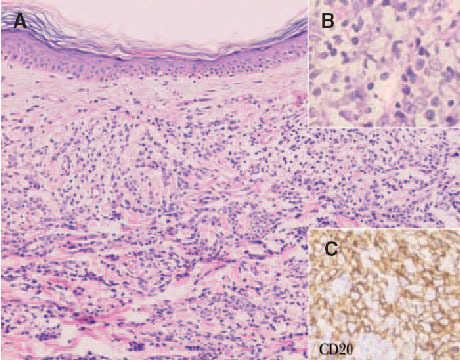
Fig. 9. A) Infiltrado difuso dérmico por células neoplásicas de gran tamaño correspondiente a un linfoma cutáneo B de células grandes de las piernas. (Hematoxilina-eosina, ×100.) B) Detalle de la marcada atipia celular. (Hematoxilina-eosina, ×200.) C) Expresión de CD20 indicando que se trata de células de estirpe B (CD20, ×200).
ENTIDADES PROVISIONALES
Linfoma cutáneo de células B intravascularEste proceso se consideraba inicialmente una proliferación vascular cutánea (32 %) con frecuente afectación del sistema nervioso central (SNC) (42 %) o, y se empleaba el término de angioendoteliomatosis maligna. Se caracteriza por acumulaciones de células B de gran tamaño de localización intravascular, que ocasionan oclusión y dilatación, y con extensión de algunas células malignas extravasculares. El inmunofenotipo de las células neoplásicas es B, con expresión de inmunoglobulinas de superficie. Excepcionalmente se han descrito casos en los que las células neoplásicas presentan un fenotipo T.
Clínicamente se caracteriza por nódulos o placas violáceas en el tronco o en las extremidades inferiores, que a veces simulan una paniculitis, con hemorragia, ulceración y necrosis acompañados de dolor y edema progresivo de la extremidad afectada. No se observan adenopatías ni visceromegalias. Son linfomas extraganglionares que tienen un comportamiento agresivo y en el momento del diagnóstico suelen presentar diseminación extracutánea (SNC). La clínica neurológica suele ser poco específica (demencia, alteraciones visuales o del habla) acompañada de sintomatología sistémica B, y otros signos y síntomas de afectación multiorgánica. Presenta una supervivencia inferior al 50 % a los 5 años, y por lo general requiere tratamiento poliquimioterápico agresivo17,46 .
Plasmacitoma cutáneoEl plasmacitoma extramedular cutáneo es una proliferación de células plasmáticas poco frecuente no relacionada en el momento del diagnóstico con un mieloma subyacente. Clínicamente se caracteriza por pápulas, nódulos o placas violáceos, solitarios o múltiples, de localización variable. Suele seguir una evolución indolente, siendo excepcional la diseminación extracutánea. Se ha estimado una supervivencia a los 5 años superior al 90 %, aunque ocasionalmente se ha descrito una evolución a mieloma9,17,47,48 . Histológicamente existe una infiltración difusa dérmica de células plasmáticas maduras y ocasionalmente multinucleadas o con macronucléolos prominentes eosinofílicos. El fenotipo de las células plasmáticas neoplásicas suele ser CD79a+, CD38+, CD138+, CD43+, CD19–, CD20–, CD22–, LCA–, con una expresión monotípica de cadenas ligeras. El diagnóstico diferencial se realiza con los LCBZM con diferenciación plasmacítica y con procesos reactivos. Debe demostrarse una expresión monotípica de las cadenas ligeras de las inmunoglobulinas.
Suelen ser tumores muy radiosensibles y de fácil manejo terapéutico.
PSEUDOLINFOMAS CUTÁNEOS
El término pseudolinfoma (hiperplasia linfoide reactiva, linfocitoma) se designa a aquellas proliferaciones linfoides benignas, que simulan tanto clínica como histológicamente un verdadero linfoma. Dependiendo del tipo celular predominante en el infiltrado, los pseudolinfomas cutáneos se dividen en pseudolinfomas cutáneos de células T y pseudolinfomas cutáneos de células B. Se considera que los pseudolinfomas cutáneos B representan una entidad clinicopatológica individualizada, por lo habitual en forma de nódulos eritematosos o violáceos. En cambio, los pseudolinfomas de células T son un grupo heterogéneo de enfermedades, más que un cuadro clínico individualizado y pueden manifestarse desde erupciones generalizadas de aspecto variable e incluso eritrodermia, a localizadas o fotolocalizadas (reticuloide actínico) e incluso en forma de placas infiltradas y nódulos.
Estos procesos parecen representar una respuesta inmunitaria exagerada frente a antígenos de diversos tipos. En la mayoría de los casos, la etiología de los pseudolinfomas cutáneos es desconocida (idiopáticos). Las causas de pseudolinfomas cutáneos de células T incluyen reacciones linfomatosas a fármacos (anticonvulsivantes, hipotensores), dermatitis de contacto linfomatoide (kathon CG), reacciones persistentes nodulares a picaduras de insecto, sarna nodular, reticuloide actínico, y el angioqueratoma pseudolinfomatoso acral. Los de células T incluyen las hiperplasias linfoides reactivas idiopáticas, hiperplasias linfoides reactivas secundarias a tatuajes, a infecciones por B. burgdorferi tras mordedura de garrapata, en el contexto de un herpes zóster o tras inyecciones con sustancias de características antigénicas.
Diagnóstico y diagnóstico diferencialEl diagnóstico diferencial entre los pseudolinfomas y los linfomas verdaderos, tanto de células T como B (especialmente los de célula de pequeño tamaño como LCBZM y LCBCF), puede ser extraordinariamente difícil. El diagnóstico definitivo se basa en la combinación de las manifestaciones clínicas, los hallazgos histopatológicos y la aplicación de técnicas inmunohistoquímicas y de estudios de clonalidad.
Histológicamente, los pseudolinfomas de células B muestran unos infiltrados nodulares simétricos bien circunscritos que predominan en las zonas superiores de la dermis y separados de la epidermis por una zona grenz. Con frecuencia el infiltrado se distribuye de forma perivascular y perianexial, aunque no suele destruir las estructuras anexiales vecinas. Habitualmente se observan folículos germinales bien formados desde un punto de vista morfológico y fenotípico. El factor más importante en la identificación de un pseudolinfoma de células B es, junto con la evolución clínica del proceso, la expresión politípica de las cadenas ligeras y de las inmunoglobulinas y la ausencia de un reordenamiento clonal de los genes de la cadena pesada y/o ligeras de las inmunoglobulinas.
Tratamiento
En los casos de pseudolinfomas de causa conocida, se hace necesaria la eliminación o el tratamiento del agente causal (sarna, B. burgdorferi , fármacos). En los pseudolinfomas idiopáticos, las lesiones pueden ser crónicas y persistentes, aunque tienden a resolverse de forma espontánea tras varios meses o años. Para las lesiones localizadas persistentes, se recomienda la extirpación quirúrgica o la criocirugía. También se han utilizado los corticoides tópicos o intralesionales. En aquellos casos con lesiones múltiples o generalizadas se han utilizado los antipalúdicos de síntesis, la foto-terapia con psoraleno y luz ultravioleta (PUVA) o fármacos citotóxicos con una respuesta variable.
CONCLUSIÓN
Los LCCB representan un grupo heterogéneo de entidades habitualmente caracterizado por una evolución clínica poco agresiva a excepción del LCBP grandes. Sin embargo, las recurrencias cutáneas no son excepcionales y sólo en algunos casos se desarrolla una enfermedad extracutánea.
Suelen ser procesos que pueden ser tratados mediante medidas terapéuticas conservadoras locales como la radioterapia o la extirpación quirúrgica. La administración intralesional de quimioterápicos, o más recientemente el empleo (local o sistémico) de rituximab, han conseguido unas tasas de respuesta variables, tanto de forma aislada como en combinación con otros tratamientos. El pronóstico depende fundamentalmente de la presencia de afectación extracutánea. En aquellos casos en los que existe una enfermedad cutánea extensa o se demuestra la presencia de afectación extracutánea puede ser necesaria la utilización de tratamientos agresivos (poliquimioterapia).
La valoración adecuada de un paciente con un posible LCCB requiere una historia clínica detallada, una exploración física minuciosa y una evaluación sistemática y detallada de los hallazgos histopatológicos, inmunofenotípicos y moleculares. Resulta fundamental la exclusión de la presencia de un proceso linfoide sistémico subyacente (linfoma secundario), mediante una estadificación y un estudio de extensión adecuado. Se aconseja realizar de forma indefinida un seguimiento clínico a todos los pacientes diagnosticados de LCCB.
Correspondencia:
Fernando Gallardo. Servicio de Dermatología. Hospital del Mar. IMAS. P.º Marítim, 25-29. 08003 Barcelona. España. FGallardo@imas.imim.es
Recibido el 8 de junio de 2004. Aceptado el 14 de junio de 2004


