







INTRODUCCIÓN
En 1956, Sneddon y Wilkinson describieron una nueva entidad bajo el nombre de dermatosis pustulosa subcórnea (DPS) (1) que posteriormente también ha sido descrita en la literatura como enferrnedad o síndrome de Sneddon-Wilkinson. Esta enfermedad, de carácter crónico y benigno, se caracteriza por la aparición sucesiva de brotes de pústulas en las flexuras, que al ir secándose evolucionan hacia la fomación de placas circinadas con nuevas pústulas en la periferia. La histología de las lesiones es característica, observándose un despegamiento a nivel subcórneo con acumulación de neutrófilos, con escasa o nula espongiosis y acantólisis.
En el año 1982, Wallach y cols. encontraron que algunos pacientes con DPS tenían una gammapatía monoclonal de tipo IgA (2) o un mieloma múltiple IgA (3-5). Sin embargo, la existencia de depósitos de IgA en la epidermis de pacientes con DPS había sido ya descrita en el año 1979 por Sneddon y Wilkinson (6) y por otros autores (7). Posteriormente se han publicado otros casos similares (8-12) y se han introducido otros términos para definir esta dermatosis: «dermatosis neutrofílica atípica con depósitos subcórneos de IgA» (13), «pénfigo foliáceo IgA» (14), etc. Algunos de estos pacientes tienen en la inmunofluorescencia indirecta anticuerpos circulantes de tipo IgA que se fijan también a las capas superiores de la epidermis.
Otra interesante asociación de la DPS es la que se ha encontrado con la piodermia gangrenosa (15-17), la enfermedad de Crohn (18) y la colitis ulcerosa (19).
Por tanto, actualmente se plantea la cuestión sobre la consideración de la DPS como una variedad de pénfigo, en concreto de pénfigo foliáceo, con depósitos intercelulares de IgA o como una dermatosis con una clínica e histología características que en algunos casos puede ser idiopática, en otros puede ser la manifestación de una nueva variedad de pénfigo denominada pénfigo IgA, y con menor frecuencia encontrarse asociada a otras patologías.
DESCRIPCION DEL CASO
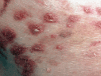
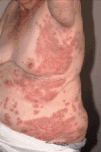
Paciente de 74 años que acudió a la consulta por unas lesiones de 5 meses de evolución que cursaban en brotes sin llegar a resolverse por completo. Se trataba de lesiones vesiculosas y pustulosas sobre una base eritematosa (Fig. 1) que evolucionaban hacia lesiones eritematodescamativas. Las lesiones se agrupaban y formaban placas anulares, circinadas, serpiginosas (Fig. 2). Inicialmente estaban localizadas en las axilas y región sacra y en las siguientes semanas aparecieron también lesiones en las nalgas, pliegues inguinales, cubitales y en el resto del tronco (Fig. 3).
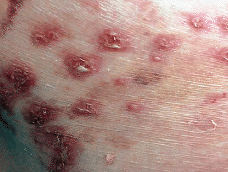
FIG. 1.--Pústulas sobre base eritematosa dispuestas de forma agrupada.
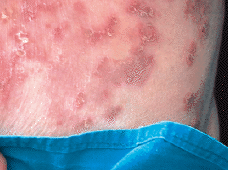
FIG. 2.--Placas eritematodescamativas con bordes circinados, serpiginosos, localizadas en axilas.
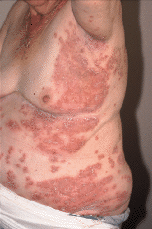
FIG. 3.--Extensión progresiva de las lesiones afectando a las axilas, cara lateral de las mamas, abdomen y flancos.
En la primera consulta se planteó el diagnóstico diferencial entre enfermedad de Sneddon-Wilkinson (dermatosis pustulosa subcórnea), psoriasis pustulosa y eritema necrolítico migratorio. Dada la disposición circinada de las lesiones, y pese a que el aspecto clínico no era el característico, descartamos que se tratase de una micosis mediante una visión de escamas con KOH y una tinción con PAS de la biopsia. Las exploraciones complementarias realizadas se encontraban dentro de la normalidad e incluyeron hemograma, bioquímica, proteinograma e inmunoelectroforesis, anticuerpos antinucleares y niveles de glucagón.
Se realizaron dos biopsias durante la evolución del proceso. En la primera, tomada de una de las lesiones axilares cuando el proceso estaba limitado a esta zona, se apreciaba un denso infiltrado inflamatorio en la porción superior de la dermis compuesto mayoritariamente por neutrófilos y linfocitos. La epidermis mostraba discreta acantosis, ortoqueratosis y una pústula de localización intracórnea, con algunas células paraqueratósicas.
La segunda biopsia se realizó 2 meses después debido a que las lesiones se habían extendido considerablemente y no existía respuesta al tratamiento. Esta biopsia mostró una epidermis acantósica con ortoqueratosis y un infiltrado en la dermis constituido por linfocitos y un gran número de neutrófilos. Los neutrófilos eran especialmente numerosos en algunas papilas y en algunos puntos se introducían en la epidermis, formando pequeños acúmulos en algunas áreas, hasta llegar por debajo de la capa córnea en donde formaban pústulas (Fig. 4). En algunas zonas la imagen era muy similar a la que ofrece una psoriasis, con ausencia de capa granulosa, acantosis, papilomatosis, paraqueratosis y acúmulos de neutrófilos en la capa córnea.
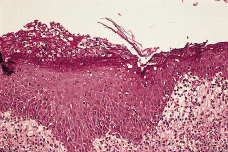
FIG. 4.--Los numerosos neutrófilos presentes en la dermis papilar se introducen en la epidermis hasta formar acúmulos por debajo de la capa córnea (pústulas subcórneas).
Con la segunda biopsia se realizó una toma para inmunofluorescencia directa (IFD) de piel perilesional que fue negativa y se tomó suero para inmunofluorescencia indirecta (IFI). Los resultados de ésta demostraron la existencia de anticuerpos IgA que se depositaban en las capas superiores de la epidermis a nivel intercelular (Fig. 5). Para determinar el antígeno concreto reconocido por estos anticuerpos se realizó un immunoblotting con extractos de epidermis humana normal separada con EDTA y un ELISA con desmogleína 1 y 3 recombinantes para detectar la existencia de anticuerpos IgG o IgA. No se encontró reactividad en ninguna de las dos pruebas. Asimismo se realizó una transfección de células COS con el cDNA de la desmocolina 1 (antígeno frente al que se generan anticuerpos en el pénfigo IgA variedad dermatosis pustulosa subcórnea); de esta manera las células COS expresan en su superficie la desmocolina 1. Con la IFI comprobamos que el suero de nuestra paciente poseía anticuerpos IgA que se unían a la superficie de las células transfectadas con un patrón granular muy llamativo (Fig. 6). Con ello confirmamos que se trataba de anticuerpos antidesmocolina 1.
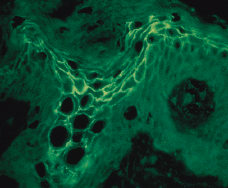
FIG. 5.--La inmunofluorescencia indirecta (IFI) realizada con el suero de la paciente demostró la existencia de anticuerpos IgA que se depositan a nivel intercelular en las capas superiores de la epidermis.

FIG. 6.--Las células transfectadas con el cDNA de la desmocolina 1 (antígeno frente al que se generan anticuerpos en el pénfigo IgA variedad dermatosis pustulosa subcórnea) expresan en su superficie este antígeno. Con una IFI comprobamos que el suero de nuestra paciente poseía anticuerpos IgA que se unían a la superficie de las células transfectadas, dando lugar a un patrón granular característico.
Iniciamos tratamiento con sulfona a dosis de 100 mg diarios; a los 15 días tuvo que ser interrumpido el tratamiento por la importante anemia que la paciente había desarrollado unida a la ausencia de respuesta clínica. Valorando las distintas alternativas que existían decidimos administrar acitretino a dosis de 25 mg al día. A las 3 semanas las lesiones empezaron a mejorar y habían desaparecido por completo a las 6 semanas (Fig. 7). El tratamiento se retiró de forma paulatina, aunque la paciente sufrió un brote menos importante de lesiones cuando tomaba 10 mg dos veces por semana, por lo que debió reinstituirse la pauta inicial de 25 mg/día que posteriormente hemos conseguido disminuir a 10 mg a días alternos.
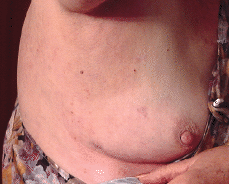
FIG. 7.--Las lesiones desaparecieron por completo 6 semanas después de iniciar tratamiento con acitretino 25 mg/día.
DISCUSIÓN
La DPS se inicia en forma de pústulas sobre una base eritematosa que van confluyendo hasta formar placas anulares o circinadas que muestran descamación y nuevas pústulas en la periferia. El centro de las placas, que es la zona en la que inicialmente existían lesiones activas, tiende a quedar hiperpigmentado. Las lesiones de forma característica se localizan en los pliegues axilares, flexuras cubitales, ingles, nalgas y abdomen. Las pústulas son estériles y los pacientes no desarrollan lesiones en mucosas.
El diagnóstico diferencial clínico de estos pacientes se plantea con el pénfigo foliáceo, el eritema necrolítico migratorio y la psoriasis pustulosa.
Histológicamente, aparte de los anteriores, el impétigo y la dermatitis herpetiforme también pueden plantear dificultades. En la dermatitis herpetiforme, el hallazgo histológico fundamental es el edema y los acúmulos de neutrófilos en las papilas de la dermis. El impétigo puede plantear grandes problemas para diferenciarlo histológicarnente de la DPS, ya que las pústulas se localizan en ambas dermatosis a nivel subcórneo, de forma que será el aspecto clínico y el cultivo microbiológico de las lesiones el que nos permita diferenciarlas. En el pénfigo foliáceo la acantolisis es más prominente que en la DPS y en ocasiones existe espongiosis eosinofílica. La IFD permite discriminar bien entre estas enfermedades debido al característico depósito de IgG intercelular en las capas altas de la epidermis que aparece en el pénfigo foliáceo. El eritema necrolítico migratorio se caracteriza por la presencia de necrosis celular en banda en el estrato espinoso, con lo que las células de esta capa adoptan una eosinofilia característica. El diagnóstico diferencial con la psoriasis pustulosa es especialmente problemático y nos referiremos a él a continuación.
El caso que hemos descrito aquí plantea una serie de cuestiones acerca de la DPS o enfermedad de Sneddon-Wilkinson. Ésta es una enfermedad que siempre ha estado sometida a controversia; de hecho, Sánchez y Ackerman ya cuestionaron en una publicación de 1979 su existencia como entidad independiente y la consideraban una forma de psoriasis pustulosa (20). Estos autores basaron sus opiniones en la llamativa semejanza histológica entre la DPS y la psoriasis pustulosa, de hecho lo único que permite diferenciarlos es la paraqueratosis que se aprecia en la psoriasis y el hecho llamativo de que los neutrófilos en la psoriasis aparecen formando pequeñas colecciones, no sólo en el estrato córneo (microabscesos de Munro), sino también en el estrato espinoso (pústulas espongiformes de Kogoj), mientras que en la DPS las pústulas se observan bajo la capa córnea y sólo algunos neutrófilos pueden verse detenidos en la capa espinosa en su migración ascendente. De todas maneras éste es un dato muy subjetivo para permitir basar el diagnóstico diferencial de las dos enfermedades únicamente en él. Por otra parte es llamativo el hecho de que la DPS responde en muchos casos a tratamientos muy efectivos para la psoriasis como los retinoides, el PUVA o los UVB.
Entre las nuevas variedades de pénfigo actualmente reconocidas se encuentra el pénfigo IgA. Esta entidad fue descrita por Wallach, Foldes y Cotenot en 1982 con el nombre de dermatosis pustular subcórnea e IgA monoclonal (2). Posteriormente se han descrito con diferentes denominaciones múltiples casos de pacientes con una clínica e histología similares y depósitos de IgA intraepidérmicos. En la actualidad se considera que existen dos variedades de pénfigo IgA: la variedad «dermatosis pustulosa subcórnea» y la variedad «dermatosis neutrofílica intraepidérrnica», con una histogía e inmunofluorescencia diferentes (20). La novedad más importante en este campo ha venido dada por el descubrimiento de los autoantígenos en ambas formas de pénfigo IgA. Los primeros estudios con immunoblotting encontraron que los anticuerpos de algunos pacientes con esta enfermedad reconocían la desmogleína 1 o la desmocolina bovinas, pero nunca las humanas. Los trabajos más recientes de Hashimoto y cols. han venido a demostrar que el autoantígeno en el pénfigo IgA con clínica e histología de DPS es la desmocolina 1 (21, 22) (proteína de la familia de las caderinas, constituyente de la porción extracelular de los desmosomas) y que la razón por la que no se detecta con el immunoblotting es porque es un antígeno conformación dependiente, es decir, que pierde su carácter antigénico y no es reconocido por los anticuerpos que se han generado in vivo al perder su conformación original, cosa que ocurre cuando se prepara un immunoblotting con epidermis humana. Por ello la mejor forma para detectarlo es utilizar células transfectadas con el DNA que codifica este antígeno (cDNA). Las celulas así transfectadas expresan en su superficie este antígeno en su conformación original, y con inmunofluorescencia indirecta se puede observar un patrón granular sobre estas células al unirlas con el suero de los pacientes con pénfigo IgA. Actualmente existe controversia sobre si la desmogleina 3, el antígeno reconocido por los autoanticuerpos IgG en el pénfigo vulgar, es también el implicado en el pénfigo IgA variedad «dermatosis pustulosa intraepidérmica» (22, 23).
Respecto al tratamiento del pénfigo IgA, la mayoría de autores señalan a las sulfonas como la primera elección (22). En la variedad dermatosis pustulosa subcórnea existen varios trabajos, algunos de ellos muy recientes, que resaltan el valor de los retinoides, tanto el acitretino como la isotretinoína (24, 25).
Nuestra opinión es que la DPS es una entidad clinicopatológica que puede responder a diferentes causas. Aunque en algunos pacientes se detectan anticuerpos IgA en la epidermis y/o circulantes, no se debe concluir, al menos con el estado actual de conocimientos, que todos los casos de DPS representan un pénfigo IgA. Las asociaciones encontradas con paraproteinemia IgA y mieloma múltiple, así como con enfermedades inflamatorias intestinales, obligan a descartar estas enfermedades en los pacientes con DPS. Finalmente en los pacientes sin anticuerpos IgA el diagnóstico diferencial con la psoriasis pustulosa anular sigue presentando dificultades importantes.
AGRADECIMIENTOS
Los estudios de inmunofluorescencia indirecta, immunoblotting, ELISA y transfección con cDNA de desmocolina 1 fueron generosamente realizados por el doctor Hashimoto, al que remitimos el suero de la paciente.


