




El dermatofibrosarcoma protuberans es el sarcoma de piel más frecuente aunque su incidencia es muy baja comparada con otros tumores cutáneos. Se presenta clínicamente en forma de placa indurada de crecimiento lento sobre la que aparecen nódulos a medida que el tumor progresa. Se localiza inicialmente en la dermis desde donde infiltra el tejido celular subcutáneo, la fascia, el músculo e incluso el hueso. La translocación COL1A1-PDGFB es específica del dermatofibrosarcoma protuberans y sirve de ayuda en el diagnóstico de determinados casos. Según la revisión de las series publicadas en la literatura, el porcentaje de recidivas con cirugía micrográfica de Mohs es mucho menor que el encontrado cuando se emplea cirugía convencional con márgenes amplios. Para casos metastásicos o en aquellos donde la cirugía pueda ser mutilante se dispone recientemente del imatinib, fármaco de la familia de los inhibitores de la tirosina quinasa.
Dermatofibrosarcoma protuberans is the most common skin sarcoma, although its incidence is very low compared with other skin tumors. It presents as a slow-growing indurated plaque on which nodules develop over time. The lesion arises in the dermis but can invade subcutaneous tissue, fascia, muscle and even bone. COL1A1-PDGFB translocation is specific to dermatofibrosarcoma protuberans, and the presence of this fusion contributes to diagnosis in certain cases. A review of the literature provides evidence that recurrence is much lower after Mohs micorgraphic surgery than after conventional wide local excision. In the case of metastatic disease or when surgery would be mutilating, another recently approved treatment is the tyrosine kinase inhibitor imatinib.
El dermatofibrosarcoma protuberans (DFSP) es un tumor cutáneo que representa los avances en Medicina que se han obtenido gracias a la biología molecular, en cuanto a diagnóstico y tratamiento. Presenta una translocación específica útil para diagnosticar determinados casos y además existe un fármaco prometedor de la familia de los inhibitores de la proteína tirosina quinasa que ha abierto una interesante posibilidad de tratamiento en los casos avanzados.
El DFSP fue descrito por primera vez en 1890 por Taylor1 como un tumor sarcomatoso que recordaba a un queloide. Sin embargo, fueron Darier y Ferrand2 en 1924 los primeros en reconocer el DFSP como una entidad propia. Un año más tarde, en 1925, Hoffman3 acuñó los términos «tumor de Darier-Ferrand o dermatofibrosarcoma protuberans» haciendo referencia a la especial tendencia de este tumor a desarrollar nódulos protuberantes en su superficie.
Actualmente se define el DFSP como un tumor cutáneo de crecimiento lento infiltrativo que presenta una alta tasa de recidivas locales pero baja capacidad metastásica. Se clasifica según la OMS4 dentro de los tumores cutáneos fibrosos, fibrohistiocíticos o histiocíticos y según Weiss y Goldblum5, en su tratado de Tumores de partes blandas, dentro de la clasificación de los tumores fibrohistiocíticos de malignidad intermedia.
Tradicionalmente se ha clasificado el DFSP dentro de las neoplasias fibrohistiocíticas, pero realmente la histogénesis del DFSP continúa siendo incierta. Según diversos trabajos el DFSP puede tener un origen fibroblástico6,7, histiocitario6,8, o neural9,10, o derivar a partir de los dendrocitos dérmicos CD 34 positivos11,12, sin embargo, muchos de esos trabajos tienen resultados contradictorios y en ningún caso se puede demostrar claramente la célula de la cual deriva el DFSP.
La expresión en el DFSP de nestina, un filamento intermedio expresado en células madre neuroectodérmicas, sugiere que el origen del DFSP sea una célula neuromesenquimal pluripotencial13–15. Esta hipótesis que explica el origen de ciertos tumores a partir de una célula madre pluripotencial mutada sería la más aceptada actualmente para el DFSP, encontrándose dicha célula madre mesenquimal, nestina positiva, en el fóliculo piloso14.
Como la mayoría de los sarcomas, el DFSP no tiene un factor de riesgo bien establecido y su etiología es desconocida. Pack y Tabah16 en 1951 sugirieron como factor etiológico del DFSP el antecedente de traumatismo local en la zona del tumor, puesto que en su serie este antecedente lo refería el 13% de los casos. Posteriormente Taylor y Helwig17 en su serie de 115 casos observan que 19 (16,5%) fueron precedidos por un traumatismo local. Desde entonces han sido muchos los casos descritos de DFSP desarrollados en la zona de un traumatismo. Este antecedente traumático que oscila del 10 al 20% de los casos podría desencadenar la aparición del tumor o ser una mera coincidencia.
Se han descrito varios casos de DFSP en mujeres en las que el tumor surge o acelera su crecimiento durante el embarazo18, incluso se encontró una alta expresividad de progesterona en 3 DFSP de mujeres embarazadas19 y se ha intentado relacionar este hallazgo con una posible etiología hormonal del DFSP pero sin resultados concluyentes. Lo que sí parece claro es que su aparición no está relacionada con la exposición solar, puesto que no se encuentran trabajos donde se demuestre esta asociación.
EpidemiologíaEl DFSP es un tumor poco frecuente. Su incidencia se ha calculado entre 0,8 y 5 casos por millón de habitantes/año20–22. Parece que la incidencia anual es superior en la raza negra que en el resto de las razas22,23. En cuanto a sexos, aparentemente existe una distribución igual entre hombres y mujeres17,24,25. Aunque las series amplias de casos muestran una mayor incidencia en hombres que en mujeres, en la revisión de 2.885 casos realizada por Criscione y Weinstock22 encuentran una incidencia ligeramente superior en el sexo femenino y en la serie de 143 casos de Martín et al.26 el 63% de los pacientes son mujeres.
Para todas las razas y sexos, la edad de mayor incidencia se encuentra entre los 30 y los 50 años22, aunque se han descrito casos congénitos y en ancianos.
La localización más frecuente del DFSP es el tronco, como muestran todas las series amplias, apareciendo en el 40 al 60% de los casos en esta localización16,17,22,26,27, sobre todo en la cintura escapular y la espalda. La segunda localización más frecuente son los miembros, viéndose afectados en un 20-30% de los casos. La cabeza y el cuello se ve afectada en el 10-15% de los casos y cuando esto ocurre suelen aparecer de manera característica en el cuero cabelludo y en la zona supraclavicular16,17,22,26,27.
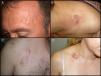
Características clínicasEl DFSP se presenta inicialmente como una pequeña placa de color de piel normal, pardusca, marrón, rosada o incluso violácea. En este estadio inicial puede pasar desapercibido por el paciente y ser confundido frecuentemente con lesiones benignas, puesto que se trata de una lesión asintomática e inespecífica. El tumor presenta un crecimiento lento y en este estadio inicial o en placa, puede adoptar 3 aspectos distintos26, el primero, tipo morfea, donde la lesión aparece como una placa indurada de color de piel normal, blanquecino o grisáceo. El segundo, tipo atrofodermia, el tumor se presenta como una placa blanda, deprimida, de aspecto atrófico, de color piel normal. El tercero, o tipo angioma, es el menos frecuente y se asemeja a lesiones vasculares como el angioma plano. A medida que el tumor crece va infiltrando en profundidad y en extensión y va desarrollando nódulos en superficie (fig. 1A, B, C y D). El tiempo en el que el tumor pasa de fase en placa o no protuberante a fase tumoral con nódulos es extremadamente variable, desde menos de un mes hasta 50 años26,27.
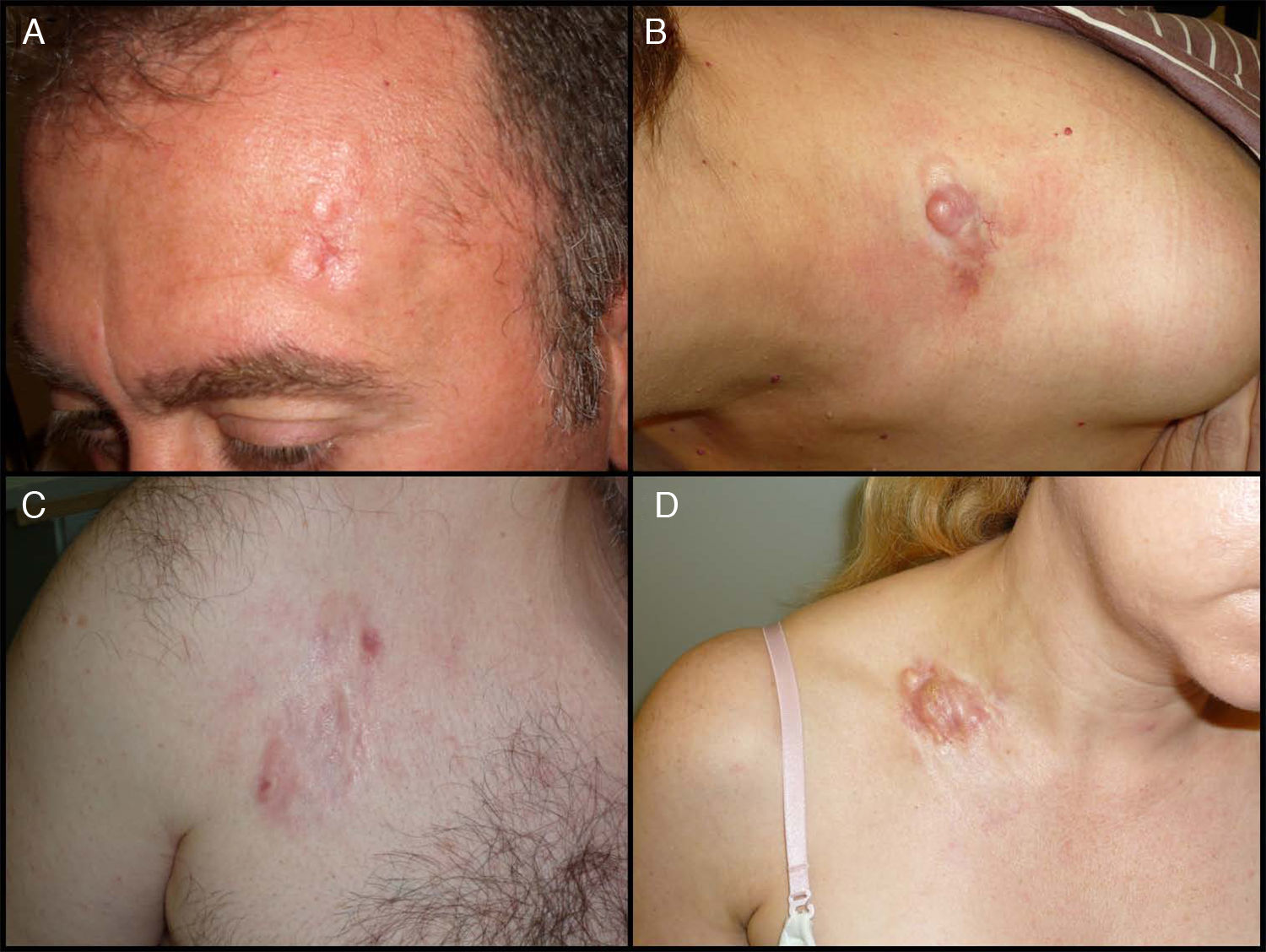
Imágenes clínicas de dermatofibrosarcoma. A. Dermatofibrosarcoma en zona frontal izquierda en forma de placa tumoral. B. Dermatofibrosarcoma en zona supraclavicular con nódulos en superficie. C. Dermatofibrosarcoma en el tórax con aspecto de placa cicatricial. D. Dermatofibrosarcoma en zona supraclavicular con nódulos en superficie.
El tamaño del tumor depende fundamentalmente del tiempo de evolución. Normalmente en el momento de la consulta suele presentar un tamaño de 1 a 5cm27 pero se han descrito casos de tamaño superior a los 20cm16.
El tumor se suele localizar en la dermis e infiltrar el tejido celular subcutáneo (TCS), por lo tanto normalmente es móvil y no adherido a planos profundos, aunque los casos con mucho tiempo de evolución pueden invadir la fascia, el músculo, el periostio y el hueso16,23,27,28.
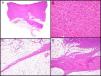
Características histopatológicasMacroscópicamente el DFSP es un tumor que se presenta como una masa única, más o menos bien delimitada en la dermis, de consistencia firme y de color amarillento o gris. En la exploración macroscópica la infiltración del TCS suele ser evidente (fig. 2A). Microscópicamente, la apariencia histológica del DFSP es la de un fibrosarcoma bien diferenciado. Está formado por una densa y uniforme proliferación de células fusiformes, con núcleo grande y elongado, con escaso pleomorfismo y baja actividad mitótica23,27. El estroma presenta cantidades variables de colágeno y capilares. Una de las características histológicas más importantes del DFSP es la disposición de estas células en fascículos entrelazados de forma irregular lo que da un patrón denominado estoriforme23,27 (fig. 2B). Con menos frecuencia, las células se pueden disponer radialmente a partir de un foco fibroso central, el denominado patrón en rueda de carro.
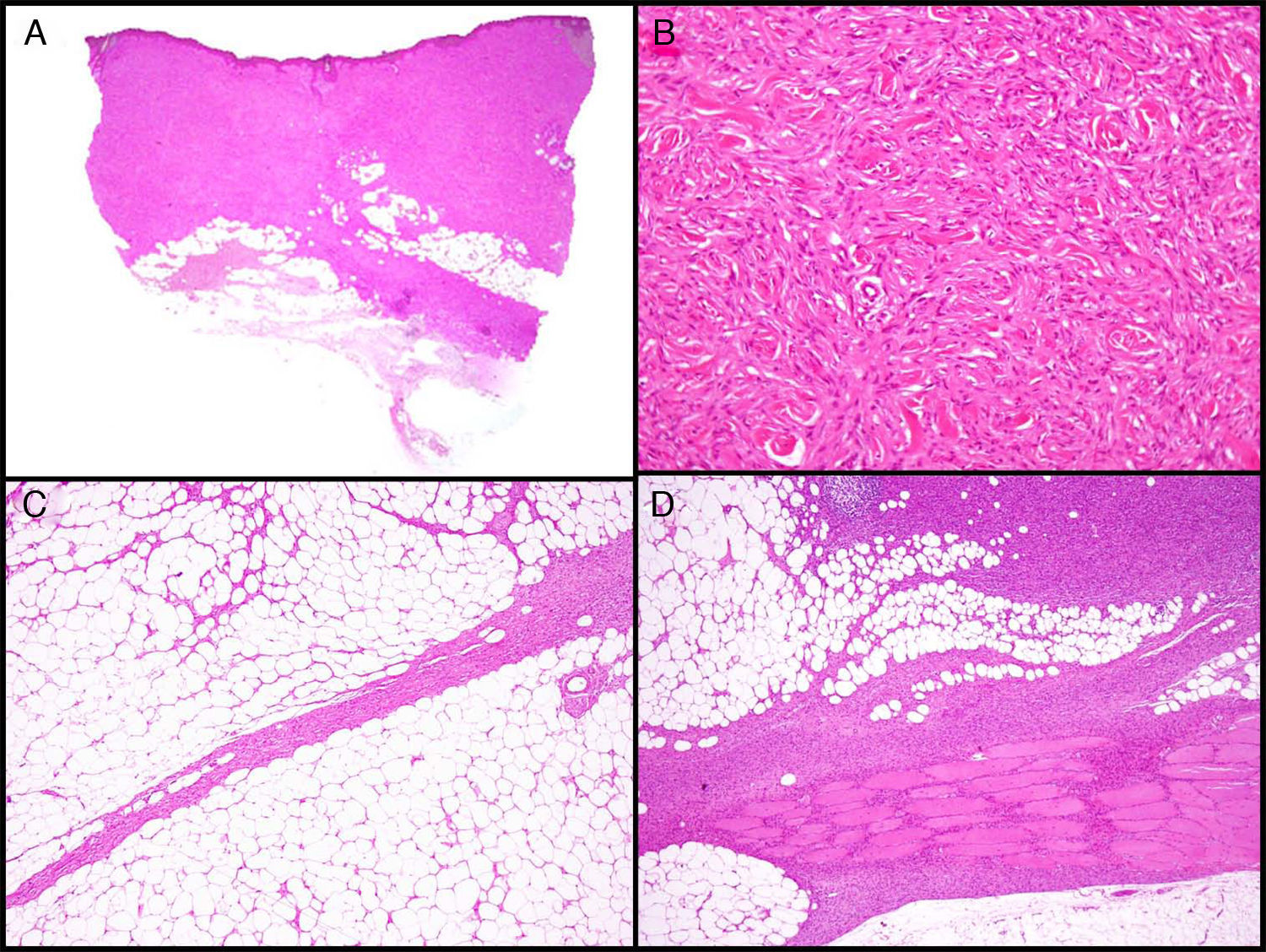
Imágenes histológicas de dermatofibrosarcoma. A. Imagen histológica de un dermatofibrosarcoma, visión panorámica (hematoxilina y eosina, 12,5x). B. Patrón estoriforme del dermatofibrosarcoma (hematoxilina y eosina, 100x). C. Infiltración del dermatofibrosarcoma en forma de prolongación digitiforme (hematoxilina y eosina, 40x). D. Infiltración muscular por dermatofibrosarcoma (hematoxilina y eosina, 40x).
La epidermis sobre el tumor suele estar adelgazada con crestas epidérmicas aplanadas. La dermis papilar normalmente está respetada, con una zona Grenz o de transición entre la epidermis y el tumor17.
La densidad celular es mucho mayor en la parte central del tumor que en la periferia, donde se emiten proyecciones digitiformes, en forma de tractos fibrosos poco celulares que infiltran el TCS, la fascia muscular, el músculo y hasta el hueso17,23,27 (fig. 2C y D). Estas prolongaciones del tumor en forma de tentáculos, que pueden llegar a gran distancia de la parte central del tumor, proporcionan al DFSP una extensión subclínica muy impredecible y pueden pasar desapercibidas en un estudio histológico convencional, lo que provoca un alto índice de recidivas incluso tras extirpaciones quirúrgicas amplias29,30. Se ha calculado que la extensión microscópica del tumor oscila de 0,3 a 12cm más allá de los márgenes macroscópicos29.
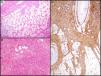
La característica fundamental del DFSP es la forma de infiltrar el TCS. Clásicamente el DFSP infiltra al TCS por los septos incluso por los lobulillos adoptando un patrón en panal de abeja (fig. 3A). En 1990 Kamino y Jacobson diferenciaron los patrones de infiltración del DFSP y del dermatofibroma (DF)31. Encontraron que el DFSP, aparte del conocido patrón en panal de abeja, infiltraba al TCS de forma más frecuente en patrón multicapa o en bandas paralelas a la superficie cutánea, dejando franjas de grasa respetada entre el tumor. Así mismo, vieron que el 60% de los casos de DFSP infiltraban en patrón en bandas paralelas, el 30% lo hacían en patrón en panal de abeja y un 10% compartían los 2 patrones. Posteriormente Zelger et al.32 confirmaron la existencia de los 2 patrones de infiltración del DFSP, aunque en su serie la mayoría presentaba el patrón en panal de abeja a diferencia de la serie de Kamino.
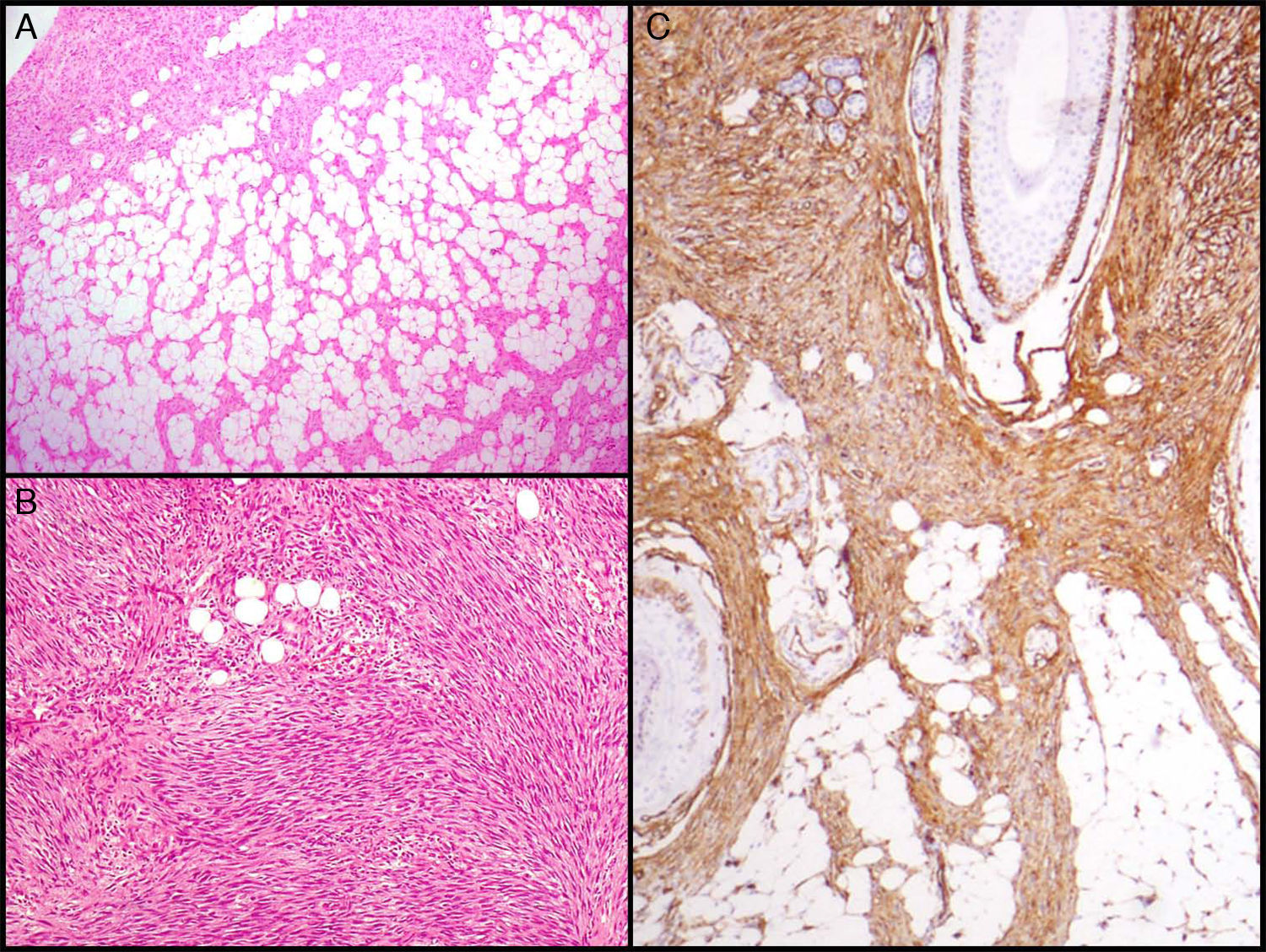
Imágenes histológicas de dermatofibrosarcoma. A. Infiltración del tejido celular subcutáneo en forma de panal de abeja (hematoxilina y eosina, 40x). B. Patrón fibrosarcomatoso de un dermatofibrosarcoma (hematoxilina y eosina, 100x). C. Tinción con CD 34 de un dermatofibrosarcoma (hematoxilina y eosina 100x).
Hasta 1957, cuando se describió un DFSP en un paciente de 5 años, se consideraba el DFSP como un tumor exclusivo de pacientes adultos. Desde entonces han sido muchos los casos descritos en la edad pediátrica y más de 30 los casos congénitos33–35.
Los DFSP congénitos presentan las mismas características inmunohistoquímicas y las mismas anomalías moleculares que el DFSP convencional, pero desde el punto de vista clínico e histológico suele haber diferencias evidentes. El DFSP congénito se suele presentar como una mácula o una placa atrófica más que como un tumor y se suele confundir con mucha frecuencia con malformaciones o tumores vasculares, morfea, atrofodermia, cicatrices atróficas o aplasia cutis33,35.
Desde el punto de vista histológico, los casos poco evolucionados pueden carecer de los hallazgos característicos del DFSP como el patrón estoriforme o la infiltración del TCS y, en tales casos, el diagnóstico se suele retrasar.
Fibroblastoma de células gigantesEn 1982, Shmookler y Enzinger36 describieron una serie de 20 casos de un raro tumor fibroblástico, que aparecía principalmente en pacientes menores de 10 años, caracterizado histológicamente por la proliferación de células fibroblásticas junto con células gigantes multinucleadas en un estroma fibromixoide al que denominaron fibroblastoma de células gigantes (FCG). Siete años más tarde, los mismos autores propusieron al FCG como la variedad infantil del DFSP37. La estrecha relación entre el FCG y el DFSP se demuestra porque ambos tumores comparten la misma clínica, histología, inmunohistoquímica y anomalías moleculares38. Sin embargo, la edad de aparición más frecuente antes de los 10 años y la presencia de células gigantes multinucleadas y un estroma mixoide en la histología caracterizan al FCG y lo diferencian del DFSP convencional.
Dermatofibrosarcoma protuberans pigmentado (tumor de Bednar)En 1957 Bednar describió un tumor, de supuesta naturaleza neural, que mostraba un patrón estoriforme junto con células fusiformes pigmentadas al que denominó neurofibroma estoriforme pigmentado39. Posteriormente, se vio que el tumor de Bednar compartía características clínicas e histológicas con el DFSP, y actualmente se le considera la variante pigmentada del mismo27. El tumor de Bednar representa del 1 al 5% de los casos de DFSP y aparece más frecuentemente en pacientes de raza negra, a diferencia del DFSP convencional27,40. Clínicamente suele ser indistinguible de un DFSP convencional, aunque, dependiendo de la cantidad de melanina que contenga el tumor, puede manifestarse como un tumor pigmentado o más frecuentemente ser un hallazgo histológico. Histológicamente el tumor de Bednar presenta como característica fundamental la presencia de una población de células dendríticas con melanina en mayor o menor proporción. Desde el punto de vista inmunohistoquímico, al igual que el DFSP convencional, es un tumor CD 34 positivo y S-100 negativo27,41.
Se han descrito casos de DFSP que recidivan en forma de tumor de Bednar42 y también la presencia de áreas fibrosarcomatosas en un tumor de Bednar metastásico. Además, el tumor de Bednar presenta alteraciones cromosómicas específicas del DFSP convencional43. Todo ello evidencia el pronóstico y la naturaleza común de este tumor y el DFSP.
Dermatofibrosarcoma protuberans atróficoEn 1985 Lambert et al.44 describieron 5 casos de DFSP con unas características clínicas que recordaban a una placa de morfea o a un carcinoma basocelular morfeiforme, puesto que presentaban un aspecto deprimido de la superficie. Como desde el punto de vista histológico eran casos claros de DFSP, propusieron el término de dermatofibrosarcoma no protuberans. Posteriormente, se describieron más casos parecidos a los de Lambert y sus colaboradores y se comenzó a utilizar el nombre de DFSP atrófico para designar a los DFSP con apariencia clínica de morfea.
Clínicamente consiste en una placa irregular, en ocasiones deprimida, de aspecto atrófico, con la presencia ocasional de telangiectasias superficiales, de color piel normal, eritematoso o pardusco45.
El DFSP atrófico se define histológicamente por la reducción de más del 50% del espesor de la dermis con relación a la dermis sana peritumoral46. Han sido descritos casos de DFSP atrófico con la translocación característica del DFSP47.
Dermatofibrosarcoma protuberans esclerosanteLa presencia de esclerosis en el seno de un DFSP es un hecho raro que parece desarrollarse de forma espontánea, sin el desencadenante de un antecedente inflamatorio o historia de radioterapia. De los 72 casos de DFSP de Díaz-Cascajo, 2 presentaban cambios escleróticos en más del 50% de la masa tumoral, lo que representaba el 3% de sus serie y confirmaba lo poco frecuente que es este hecho en el DFSP48.
Desde el punto de vista histológico, en las áreas de esclerosis las células neoplásicas son gradualmente reemplazadas por tejido esclerótico sin una reducción significativa del espesor tumoral, como sucede en el DFSP atrófico. La cantidad de colágeno en estas áreas se correlaciona con la pérdida de la celularidad tumoral49. También han sido descritos casos donde las zonas escleróticas representan nódulos tumorales50. Las áreas escleróticas son parcialmente positivas al CD 34, hecho que se correlaciona con la disminución de células tumorales que permanecen embebidas en el estroma esclerótico.
Para Díaz-Cascajo48, la producción excesiva de colágeno por las células tumorales, sin la coexistencia de inflamación, y la presencia de transición entre las típicas áreas de DFSP y las áreas escleróticas pueden representar un signo de involución. En este sentido, los 3 casos descritos por Hattori50 corresponden a DFSP de larga evolución, lo que podría dar sentido a la teoría de Díaz-Cascajo.
Dermatofibrosarcoma protuberans mioideLa presencia de áreas de diferenciación mioide en el DFSP es un hecho raro que fue descrito por primera vez en 1996 por Calonje y Fletcher51. Describieron 5 casos de DFSP comunes o DFSP con componente fibrosarcomatoso con presencia de haces y nódulos dispersos y confluentes de células fusiformes eosinófilas, de citoplasma bien delimitado y núcleo vesicular muy semejantes a células musculares lisas o miofibroblastos. Dichas áreas mioides eran CD 34 negativas pero sí se teñían para actina. Estas zonas aparecían tanto en la superficie como en la profundidad del tumor y no se localizaban específicamente en proximidad ni al músculo erector pili ni a las paredes vasculares, sino que lo hacían al azar, lo que indicaba que eran zonas propias del tumor. Sin embargo, muchos autores piensan que estas áreas mioides no son una verdadera diferenciación del DFSP sino más bien un fenómeno reactivo representado en forma de miofibroblastos estromales hiperplásicos52,53.
Independientemente del significado exacto de este hallazgo, la presencia de áreas de diferenciación mioide en el DFSP es un hecho raro y suele acontecer, sobre todo, en casos de DFSP con componente fibrosarcomatoso51.
Dermatofibrosarcoma protuberans mixoideLa variedad mixoide del DFSP fue descrita por primera vez en 1983 por Frierson y Cooper54. Posteriormente se han descrito varios casos y una serie amplia de 23 casos en 2007 por Reimann et al.55. En todos los casos coinciden en destacar del DFSP mixoide que es muy poco frecuente.
Desde el punto de vista clínico y pronóstico se comporta de manera similar al DFSP convencional, con la única peculiaridad de que parece localizarse con más frecuencia en miembros que en el tronco55. Histológicamente el tumor está compuesto por células fusiformes o estrelladas con citoplasma pálido, eosinófilo dispuestas en lóbulos. La presencia de gran cantidad de material mixoide en el estroma, que por definición está presente en mas del 50% del tumor55, conlleva que el patrón estoriforme suela ser menos aparente. Las mitosis son escasas y no suele haber mucho pleomorfismo. De manera característica presenta numerosos vasos ramificados de paredes finas. La positividad de las células para el CD 34, que se ve en la mayoría de los casos, y el patrón de infiltración en panal de abeja suelen ser los criterios diagnósticos más importantes puesto que en la mayoría de los casos el diagnóstico histológico es complejo.
Dermatofibrosarcoma protuberans con componente fibrosarcomatosoEn 1951 Penner describió un caso de DFSP metastásico que contenía áreas de fibrosarcoma56. En los años sucesivos se han publicado muchos casos y series de DFSP con componente fibrosarcomatoso (DFSP-FS). Wang et al.57 realizaron un estudio en el que analizaron por separado la zona de DFSP y la zona de FS en 6 DFSP-FS y encontraron las mismas alteraciones moleculares en las 6 muestras de DFSP y en 5 de las de FS, lo que apoya la histogénesis común de estos 2 componentes. La presencia de áreas fibrosarcomatosas en los DFSP se observa en un 7-17% de los casos según las series40,58,59. Desde el punto de vista clínico y epidemiológico el DFSP-FS no difiere del DFSP convencional. Histológicamente se caracteriza por presentar, en el seno de un DFSP, áreas histológicamente indistinguibles de un fibrosarcoma. Estas zonas se caracterizan por una densa proliferación de células fusiformes dispuestas en largos fascículos que se cruzan formando un patrón en espina de pescado58. Las áreas fibrosarcomatosas pueden ocupar desde un 5 a un 90% del tumor, pero según Weiss y Goldblum se considera que un DFSP presenta componente fibrosarcomatoso cuando este ocupa por lo menos un 5-10% del tumor5 (fig. 3B). La tinción inmunohistoquímica con CD 34 en las áreas fibrosarcomatosas suele ser de menor grado o estar ausente en comparación con las áreas de DFSP contiguas. Por el contrario, se ha demostrado que en las áreas fibrosarcomatosas existe una sobreexpresión de la p-5359 y un alto índice mitótico.
Características inmunohistoquímicasFilamentos intermediosLa vimentina es una proteína que se expresa en todas las células mesenquimales; por lo tanto, prácticamente todos los sarcomas, incluido el DFSP son vimentina positivos60.
La nestina es un filamento intermedio que se describió por primera vez como marcador de células madre neuroectodérmicas y posteriormente se identificó también en células madre mesenquimales de médula ósea, pulmón, músculo y páncreas11. Recientemente se ha demostrado su utilidad en el DFSP, sobre todo para diferenciarlo del DF. Mori et al.15 comparan la tinción con nestina y otros marcadores inmunohistoquímicos en 16 casos de DFSP y en 30 de DF. Obtienen positividad para la nestina en el 94% de los DFSP y solamente en el 13% de los DF. En otros trabajos más recientes se obtienen resultados más contundentes todavía, puesto que encuentran intensa positividad en todos casos de DFSP13,14.
CD 34En la piel normal, el CD 34 se expresa en las células endoteliales, en las células fusiformes perifoliculares, en las células fusiformes perivasculares y perianexiales, y en las células dendríticas intersticiales de la dermis reticular61,62.
El CD 34 se consideraba un marcador relativamente específico para neoplasias de origen vascular, sin embargo, en 1992 Aiba et al.62 estudian la expresión del CD 34 en una serie de tumores fibrohistiocitarios como el DF, el DFSP, cicatriz hipertrófica y queloide, y encontraron que el único tumor que lo expresaba era el DFSP. Posteriormente Kutzner encuentra hallazgos similares63. En 1994 Cohen et al.64 revisan todas las serie publicadas hasta entonces (9 series con 96 casos) y calculan que el 88% de los DFSP son CD 34 positivos. Desde entonces han sido varias las series descritas donde se muestran los resultados de la inmunotinción con CD 34 y se encuentra positividad en el 92-100% de los casos65–67; por lo tanto, es un marcador muy útil para diferenciar el DFSP de otros tumores fibrohistiocitarios, sobre todo del DF (fig. 3C).
A pesar de que la mayoría de los casos de DFSP expresa el CD 34, la positividad del CD 34 se ha descrito en multitud de tumores, tanto benignos como malignos, como tumor miofibroblástico inflamatorio, tumor fibroso solitario, fibroma esclerótico, fibromixoma acral superficial, sarcoma de Kaposi, neurofibroma, perineurioma, o melanoma61.
Factor XIIIaEl factor XIIIa es una proteína tetramérica que se expresa, en la piel normal, en los dendrocitos dérmicos de la dermis papilar, especialmente alrededor de los vasos superficiales67. En 1989 Cerio et al.68 encuentran positividad para el factor XIIIa en 30 casos de DF, en comparación con 16 casos de DFSP donde hallan ausencia o muy escasa inmunotinción, sugiriendo que este marcador podía ser interesante para diferenciar estos 2 tumores. En la mayoría de los estudios se obtiene positividad en el 90-95% de los DF y, además, la expresión se obtiene en la mayoría de las células y de manera intensa, a diferencia del DFSP donde únicamente son positivos el 10-15% de los casos64,67,69.
Otros marcadores inmunohistoquímicosEl diagnóstico diferencial del DFSP se establece frecuentemente con el DF y para ello, el CD 34 (positivo en la mayoría de los DFSP y hasta en el 25% de los DF69) y el factor XIIIa (negativo en la mayoría de los DFSP y positivo en casi todos los DF) son los marcadores más utilizados; sin embargo, se ha descrito la utilidad de otros marcadores.
En 2004 West et al.70 estudiaron la expresión de apolipoproteína D (APO D) en 421 tumores de partes blandas y encontraron que el tumor que más expresaba este marcador era el DFSP a diferencia de los DF comunes que resultaron todos negativos. Posteriormente otros trabajos confirman que la APO D puede ser interesante para diferenciar el DFSP del DF.
Otro marcador inmunohistoquímico que ha sido estudiado en el DFSP es la estromelisina 3. En dichos trabajos los resultados muestran mayor grado de expresión en el DF que en DFSP con diferencias más o menos llamativas según los estudios71.
La mayoría de los trabajos coinciden en que el DFSP tiene un índice proliferativo bajo, demostrado tanto con Ki-67 como con MIB-159,72.
La expresión inmunohistoquímica de la p53 suele ser negativa o débilmente positiva en el DFSP y se asocia con más frecuencia al DFSP-FS59. Sin embargo la p53 puede ser de útil en la distinción del DFSP y el DF puesto que este último no suele expresarla.
Citogenética y biología molecular del dermatofibrosarcoma protuberansLa citogenética del DFSP comenzó en 1990 con las primeras descripciones de Bridge et al.73 y de Mandahl et al.74 con 2 casos de DFSP en los que aparecía un cromosoma en anillo supernumerario indeterminado. Posteriormente, el análisis con hibridación in situ con fluorescencia (FISH) demostró que el anillo del DFSP contenía secuencias del cromosoma 1775. La combinación de las técnicas de FISH e hibridación genómica comparada demostraron la implicación del cromosoma 22 en la formación del cromosoma en anillo76. Desde entonces se considera algo característico del DFSP la combinación de secuencias del cromosoma 17 y 22 contenidos en un cromosoma en anillo supernumerario derivado del cromosoma 2243.
Simon et al.77, con técnicas de FISH y de biología molecular, identificaron el punto de fusión exacto y comprobaron que en él estaban identificados 2 genes (el gen del factor de crecimiento beta derivado de las plaquetas [PDGFB] [22q13.1] y el gen del colágeno I alfa 1 [COL1A1] [17q.22]) y que daban lugar a un gen quimérico.
En todos los casos publicados de DFSP con el gen de fusión COL1A1-PDGFB la localización del punto de corte del gen PDFGB es constante en el exón 2, sin embargo, la localización del punto de corte del gen COL1A1 es muy variable. En la literatura se describe la implicación de 27 exones diferentes, siendo los más frecuentemente implicados el 24, el 29 y el 32, y no parece haber ninguna trascendencia en cuanto a la clínica, la histología o el pronóstico en función del punto de corte del COL1A158.
Los distintos estudios sugieren que el resultado de la translocación con el gen de fusión COL1A1-PDGFB en el DFSP produce una proteína quimérica que posteriormente es procesada para originar PDGFB maduro y plenamente funcional.
Por lo tanto, la t(17;22) en el DFSP da lugar a una activación del receptor del PDGFB por medio de una producción autocrina y paracrina de un ligando funcional que se traduce en una señal mitogénica crónica, capaz de inducir la transformación neoplásica.
Hasta la fecha, el DFSP es el único tumor en el cual se ha demostrado una alteración somática de los genes COL1A1 y PDFGB. Además de la translocación t(17;22) propia del DFSP, se han descrito otras anomalías cromosómicas, numéricas y estructurales en el DFSP. De estas, la trisomía 8 es la anomalía más frecuente ya que aparece en un tercio de los DFSP cariotipados76.
Características radiológicasSe han descrito casos de DFSP estudiados con radiografías convencionales, con tomografía axial computarizada e incluso con arteriografía, pero en ninguno se ha obtenido información precisa o no existen características específicas con estas pruebas en el DFSP que ayuden a diagnosticarlo o a precisar su localización.
Según la serie más larga de resonancia magnética (RM) en DFSP78, esta tiene mayor sensibilidad y especificidad que la palpación clínica para determinar afectación en profundidad; sin embargo, la RM pierde fiabilidad en tumores localizados en la cabeza y el cuello.
Tratamiento del dermatofibrosarcoma protuberansLa extirpación quirúrgica completa es el tratamiento de elección para el DFSP27,29. Es preciso tener en cuenta que el modo de crecimiento del DFSP, mediante proyecciones digitiformes, lo convierte en un tumor muy asimétrico, cuya extensión subclínica puede llegar a gran distancia del centro del tumor29. Estas proyecciones en forma de tentáculos en la periferia del tumor pueden pasar desapercibidas incluso en un estudio histológico convencional y explican el alto índice de recurrencias locales que presenta el DFSP23.
El empleo de cirugía convencional o de cirugía micrográfica de Mohs (CMM) para el tratamiento del DFSP, es un debate que persiste hoy en día.
Cirugía convencional en el dermatofibrosarcoma protuberansExisten muchas series en la literatura de DFSP tratados con cirugía convencional, sin embargo, la forma de extirpar los DFSP varía considerablemente de unos trabajos a otros y, por lo tanto, es complicado extraer conclusiones concretas acerca del empleo de la cirugía convencional en el DFSP. En la tabla 1 se exponen las series descritas en la literatura de al menos 10 casos de DFSP tratados con cirugía convencional. Existen 38 series de más de 10 casos de DFSP extirpados con cirugía simple, desde 1951 a 2011 que incluyen un total de 1.782 pacientes16,17,20,24,25,30,79–110. Al analizar el margen de resección, en la mayoría de las series utilizan el término «exéresis amplia» sin concretar la medida en milímetros o centímetros de piel clínicamente sana que extirpan; además, en muchos casos los tumores se extirpan varias veces hasta conseguir márgenes histológicamente negativos102,105,109. En otras series utilizan un margen de seguridad que puede ser de 1 hasta 5 cm y sí que existe una tendencia a conseguir un menor número de recidivas en los casos extirpados con mayores márgenes de seguridad88,101. Por otra parte, tampoco se especifica en la mayoría de las series la inclusión o no de la fascia muscular subyacente en la exéresis de los DFSP. Pese a todo, se podría calcular que el DFSP recidiva hasta en un 60% de los casos que utilizan cirugía con márgenes amplios, sin especificar el margen empleado ni la inclusión o no de la fascia muscular en la exéresis. Solamente en 9 de las 38 series no obtienen ninguna recidiva y en más de la mitad de las series las recidivas superan el 20% de los casos.
Series de 10 casos o más de dermatofibrosarcomas tratados con cirugía convencional
| Cita/año | N° de pacientes | Margen en cm | Seguimiento | Recurrencias |
| Gentele, 1951 | 38 | - | - | 16 (42%) |
| Pack, 1951 | 39 | Amplia | 6 meses-20 años | 8 (21%) |
| Taylor, 1962 | 98 | Amplia | 1-17 años | 48 (49%) |
| Burkhardt, 1966 | 21 | Amplia | > 5 años | 7 (33%) |
| Longhin, 1967 | 44 | - | 1-11 años | 14 (32%) |
| McPeak, 1967 | 82 | 3 | 3-15 años | 8 (10%) |
| Tamoney, 1971 | 12 | Amplia | 1-30 años | 3 (25%) |
| Bendix-Hansen, 1983 | 19 | 1-3 | 4-13 años | 8 (42%) |
| Barnes, 1984 | 15 | - | 1-23 años | 8 (53%) |
| Waldermann, 1985 | 13 | 4-5 | 1-7 años | 3 (23%) |
| Petoin, 1985 | 96 | 4 | 1-15 años | 6 (6%) |
| Roses, 1986 | 48 | >2 | >3 años | 16 (33%) |
| Chattopadhyay, 1986 | 10 | Amplia | 5-7 años | 6 (60%) |
| Rutgers, 1992 | 19 | >2 | 2-28 años | 8 (42%) |
| Brabant, 1993 | 14 | 5 | 1-5 años | 0 (0%) |
| Koh, 1995 | 19 | - | > 3 años | 5 (26%) |
| Gloster, 1996 | 39 | Amplia | 1 mes-14 años | 5 (13%) |
| Arnaud, 1997 | 107 | 5cm | 5 años | 2 (1.86%) |
| Hass, 1997 | 21 | Amplia | 1-10 años | 7 (33%) |
| Bowne, 2000 | 159 | Amplia | 5 años | 34 (21%) |
| Joucdar, 2001 | 81 | 5 | 5 años | 14 (17,3%) |
| Khatri, 2003 | 24 | Amplia | 4.5 años | 0 (0%) |
| Chang, 2004 | 60 | 3 | 5 años | 10 (16,7%) |
| Tan, 2004 | 10 | Amplia-3cm | 2-9 años | 0 (0%) |
| DuBay, 2004 | 43 | 1-2 | 4 años | 0 (0%) |
| Fiore, 2005 | 218 | Amplia | 10 años | 8 (4,3%) |
| Behbahani, 2005 | 34 | 3 | 5 años | 0 (0%) |
| Szollosi, 2005 | 28 | Amplia | 4-26 años | 6 (21,4%) |
| Ruiz-Tovar, 2006 | 21 | - | 3 años | 6 (28,5%) |
| Monnier, 2006 | 66 | 1-5 | 9 años | 17 (27%) |
| Popov, 2007 | 40 | 3 | 3 años | 0 (0%) |
| Paradisi, 2008 | 38 | 2-5cm | 5 años | 5 (13%) |
| Yu, 2008 | 14 | 3-5cm | 32-133 meses | 0 (0%) |
| Bague, 2008 | 15 | Amplia | 3-19 meses | 0 (0%) |
| Edelweiss, 2010 | 13 | Amplia | 2-444 meses | 7 (53%) |
| Meguerditchian, 2010 | 28 | 1-3 | 4 años | 1 (3,6%) |
| Archontaki, 2010 | 16 | Amplia | 3 años | 0 (0%) |
| Erdem, 2011 | 120 | Amplia | 10,2 | 38 (31,7%) |
El primer autor en publicar el empleo de la CMM en el DFSP fue el propio Dr. Mohs en 1978111. Desde entonces han sido muchos los casos aislados y las series publicadas que demuestran la utilidad de la CMM en el tratamiento del DFSP y la proponen como tratamiento de elección112. Se han publicado 26 series de 10 casos o más de DFSP tratados con CMM29,30,78,96,105,109,113–133 (tabla 2). Según las 27 series de DFSP tratados con CMM, las recidivas con esta técnica son del 0-8,3%, porcentajes muy inferiores a los encontrados cuando se emplea cirugía con márgenes amplios. Además, en 22 de las 27 series no encuentran ninguna recidiva con seguimientos superiores a los 2 años. Por otra parte, la CMM proporciona el verdadero y ajustado margen exclusivo de cada caso a diferencia de la extirpación convencional con márgenes amplios que puede ser incompleta y al mismo tiempo puede extirpar una gran cantidad de tejido sano no tumoral.
Series de 10 casos o más de dermatofibrosarcomas tratados con cirugía de Mohs
| Cita/año | N° de pacientes | Seguimiento | Recurrencias |
| Hobbs, 1988 | 10 | 1,25-7,6 años | 0 |
| Breuninger, 1994 | 23 | 5 años | 0 |
| Parker, 1995 | 20 | 3 meses-8,75 años | 0 |
| Gloster, 1996 | 15 | 5 meses-8 años | 1(6,6%) |
| Garcia, 1996 | 16 | 4,4 años | 0 |
| Dawes, 1996 | 24 | 5,1 años | 2 (8,3%) |
| Ratner, 1997 | 58 | 4,8 años | 1(1,7%) |
| Haycox, 1997 | 10 | 3,4 años | 0 |
| Clayton, 2000 | 11 | 2 años | 0 |
| Huether, 2001 | 33 | 3,8 años | 1(3%) |
| Ah-Weng, 2002 | 21 | 4 años | 0 |
| Nouri, 2002 | 20 | 4,7 años | 0 |
| Wacker, 2004 | 22 | 4,5 años | 0 |
| Snow, 2004 | 29 | 5 años | 0 |
| Sei, 2004 | 10 | 2,2 años | 0 |
| DuBay, 2004 | 11 | 5,2 años | 0 |
| Thomas, 2007 | 35 | 3,3 años | 0 |
| Gattoni, 2007 | 31 | 3 años | 0 |
| Cecchi, 2007 | 10 | 4,1 años | 0 |
| Häfner, 2008 | 70 | 5 años | 1 (1,4%) |
| Paradisi, 2008 | 41 | >5 años | 0 |
| Hancox, 2008 | 25 | 8,4 años | 0 |
| Nelson, 2008 | 44 | 3,3 años | 0 |
| Meguerditchian, 2010 | 20 | 3,3 años | 0 |
| Roh, 2010 | 11 | 2,1 años | 0 |
| Tan, 2011 | 35 | 2,4 años | 0 |
| Serra-Guillén, 2011 | 43 | 2,9 años | 0 |
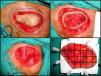
La variante más aceptada es la denominada «slow Mohs» o Mohs diferido125. Esta se realiza de la siguiente manera (fig. 4A, B, C y D). En primer lugar se realiza un debulking o extirpación ajustada del tumor o de la cicatriz en el caso de los tumores extirpados recientemente con márgenes afectados. Posteriormente se toma el primer estadio de 0,5 a 1cm de piel clínicamente sana, incidiendo el bisturí con un ángulo de 45° y llegando en profundidad hasta la totalidad del tejido celular subcutáneo e incluyendo las capas más superficiales de la fascia muscular. Antes de retirar este primer estadio, se referencia la pieza con puntos de seda y se realiza una fotografía. El debulking se procesa de forma convencional, se fija en formol y se incluye en parafina, con posterior obtención de cortes paralelos teñidos con hematoxilina y eosina. El primer estadio de Mohs se remite sobre una placa de espuma de poliuretano sujetado con agujas, para mantener su forma y evitar la retracción, y todo ello en un cubo con formol. También se remite al patólogo la fotografía que se había realizado con la pieza todavía en el propio paciente. El patólogo separa en primer lugar los márgenes laterales con cortes que incluyen desde la epidermis hasta el fondo de la pieza y posteriormente separa el fondo realizando cortes horizontales. A todas las piezas obtenidas se les asigna un número y se reproduce todo el proceso en la fotografía. Los cortes se tiñen con hematoxilina y eosina y se estudian en el microscopio buscando algún foco de DFSP. En el caso de encontrar zonas dudosas o difíciles de interpretar, se tiñe el mismo corte con la tinción inmunohistoquimica de CD 34. Si tras estudiar todos los márgenes no se halla ningún foco de DFSP se procede al cierre del defecto. Si se encuentra algún margen afectado por DFSP se continúa la extirpación utilizando la fotografía como referencia para conocer exactamente el margen afecto. Este procedimiento se realiza hasta obtener la negativización de todos los márgenes, tras lo cual se procede al cierre definitivo del defecto. La ventaja que aporta la variante diferida o slow Mohs es que los cortes tienen mucha mayor calidad y es mucho más fácil de diagnosticar la presencia de focos de DFSP, a diferencia de la CMM convencional que al procesar la pieza en congelado proporciona un corte histológico más difícil de interpretar. Por otra parte, el tiempo empleado en la variante diferida es mucho mayor que en la CMM convencional.
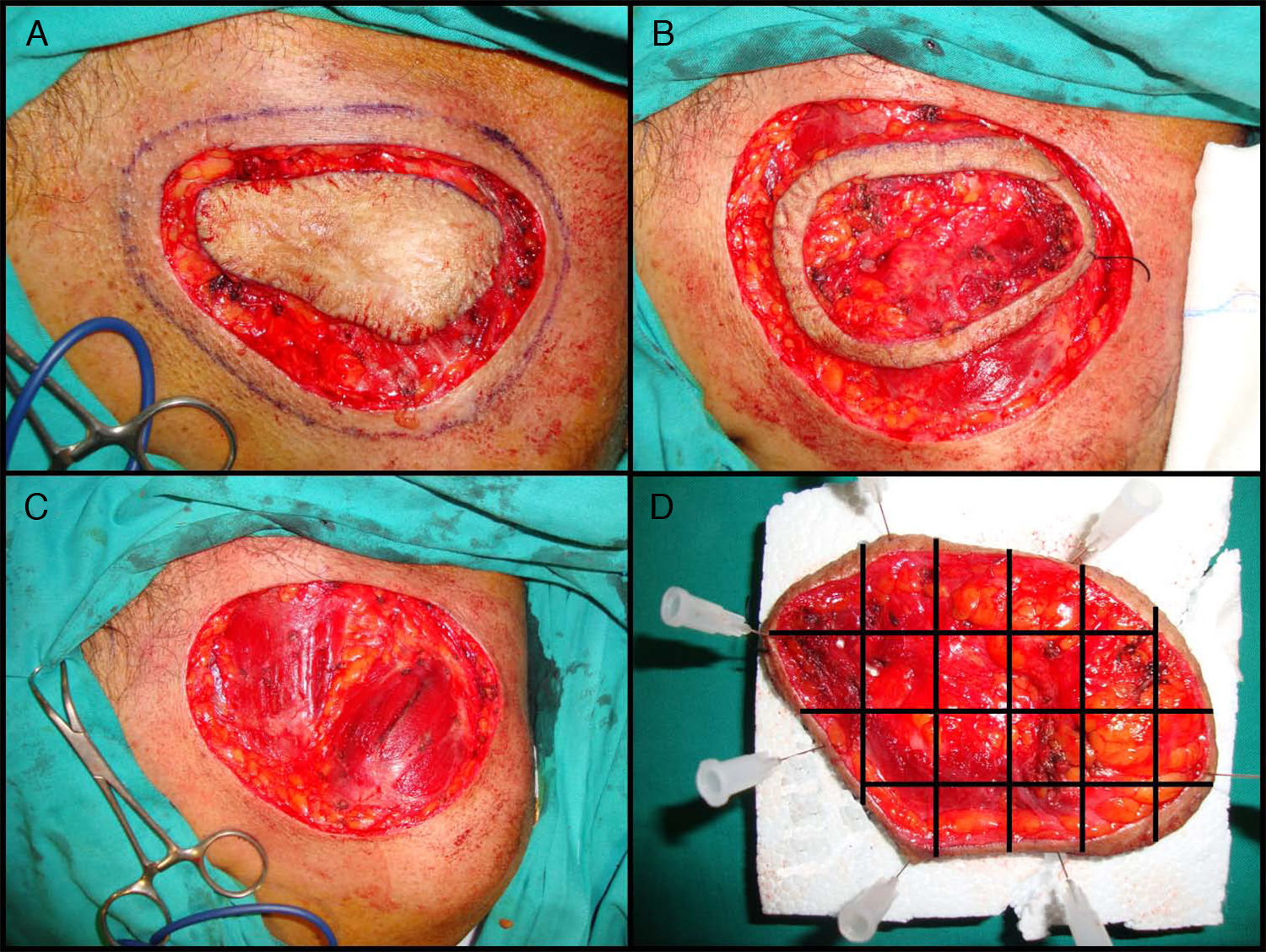
Cirugía de Mohs diferida en el dermatofibrosarcoma protuberans. A. Debulking o extirpación ajustada del tumor. B. Primer estadio de Mohs. Anillo cutáneo de piel clínicamente sana con referencia. C. Defecto tras el primer estadio. Exposición del plano muscular. D. División de la pieza por el patólogo.
La identificación de una expresión desregulada del receptor del PDGFB como resultado de la translocación t(17;22) llevó a la hipótesis de que los inhibidores de la proteína tirosina quinasa presentes en dicho receptor, como el imatinib, podrían tener una actividad en el DFSP. El imatinib se fija de forma competitiva en el receptor PDGF y bloquea su actividad tirosina quinasa. Después de los estudios preclínicos con resultados favorables, se han publicado varios trabajos con buenas respuestas del imatinib en DFSP metastásicos o localmente avanzados108,134–153. Se ha empleado de manera neoadyuvante, previo a la extirpación quirúrgica, con una reducción del tamaño tumoral que oscila del 19% hasta la respuesta clínica completa108,134-153 (tabla 3). Sin embargo, es necesario ser crítico con los trabajos donde se han obtenido respuestas completas para poder extender el uso de imatinib en el DFSP. En lo que respecta a los 4 casos con respuesta completa publicados por McArthur138, además de su escaso periodo de seguimiento, solamente realizan estudio histológico en 2 casos. En este sentido, el DFSP tras el tratamiento con imatinib proporciona una imagen histológica característicamente muy poco celular con abundante colágeno hialinizado, hecho que puede producir la falsa interpretación de ausencia tumoral138,141. Por lo tanto, para demostrar o descartar la persistencia tumoral después del tratamiento con imatinib en un DFSP aparentemente resuelto tras el estudio histológico convencional, debe estudiarse la existencia de la translocación COL1A1- PDGFB. A pesar de todo, según todos los estudios publicados, el imatinib parece beneficioso como tratamiento neoadyuvante en casos localmente avanzados o como tratamiento paliativo en casos metastásicos.
Casos publicados de dermatofibrosarcoma protuberans tratados con imatinib
| Cita/año | Caso | Duración de tratamiento | Respuesta | Seguimiento |
| Rubin, 2002 | Metastásico | 4 meses | Parcial (reducción 75% | - |
| Maki, 2002 | Metastásico | 4 semanas | Parcial | Muerte |
| Metastásico | 2 meses | Casi completa | ||
| Mizutani, 2004 | Metastásico | 3 meses | Parcial | - |
| Labropoulos, 2005 | Metastásico | 20 meses | Completa | 20 meses |
| McArthur, 2005 | Local. Avanzado | 698 días | Parcial | 845dias |
| Local. Avanzado | 62 días | Parcial | 699 días | |
| Local. Avanzado | 141 días | Parcial | 572 días | |
| Local. Avanzado | 457 días | Completa | 536 días | |
| Local. Avanzado | 139 días | Parcial | 258 días | |
| Local. Avanzado | 188 días | Completa | 267 días | |
| Local. Avanzado | 146 días | Completa | 225 días | |
| Local. Avanzado | 88 días | Completa | 88 días | |
| Metastásico | 198 días | Parcial | 383 días | |
| Metastasico | 21 días | Estable | 32 días | |
| Price, 2005 | Local. Avanzado | 23 semanas | Parcial | - |
| Mehrany, 2006 | Local. Avanzado | 24 meses | Parcial | 18 meses |
| Savoia, 2006 | Local. Avanzado | 11 meses | Parcial | 6 meses |
| Kasper, 2006 | Metastásico | 4 meses | Parcial | - |
| Wright, 2007 | Local. Avanzado | 5 meses | Parcial | 16 meses |
| Kim, 2007 | Metastásico | - | Completa | 1 año |
| Heinrich, 2008 | 12 casos (sin especificar) | 3-12 meses | 4 Completa6 Parcial1 Progresión1 Desconocido | 23,9 meses |
| Thomison, 2008 | Local. Avanzado | 1 año | Parcial | - |
| Lemm, 2008 | Local. Avanzado | 3 meses | Parcial (Reducción 60% | 6 meses |
| Llombart, 2009 | Metastásico | 4 meses | Parcial | Muerte |
| Local. Avanzado | 1 año | Parcial (reducción 50%) | 1 año | |
| Han, 2009 | Local. Avanzado | 3 meses | Parcial (reducción 19%) | 1,5–4 años |
| Local. Avanzado | 3.5 meses | Parcial (reducción 45%) | ||
| Local. Avanzado | 7 meses | Parcial (reducción 62%) | ||
| Local. Avanzado | 3 meses | Parcial (reducción 22%) | ||
| Rutkowski, 2010 | 24 casos«7 metastásicos» | 248-328 días | 9 Parcial8 Estable4 Progresión3 No evaluable | 2,6 años |
| Gooskens, 2010 | Local. Avanzado | 6 meses | Parcial (reducción 30%) | 3,5 años |
| Local. Avanzado | 12 meses | Parcial | 3 años | |
| Local. Avanzado | 6 meses | Parcial | 6 meses | |
| Kerob, 2010 | 25 casos«20 primarios»«5 recidivas» | 2 meses | ParcialReducción <30% en 16 casos>30% en 9 casos | - |
| Edelweiss, 2010 | Local. Avanzado | - | - | 72 meses |
| Stacchiotti, 2011 | Local. Avanzado | 4 meses | Parcial | 5 meses |
| Metastásico | 4 meses | Parcial | Muerte | |
| Metastásico | 4 meses | Parcial | Muerte | |
| Metastásico | 4 meses | Parcial | Muerte | |
| Rutkowski, 2011 | 15 casos«9 local avanzado»«6 metastásicos» | - | 10 Parcial2 Estables3 Progresión | 16 meses |
El papel de la radioterapia en el manejo del DFSP ha sido poco estudiado y parece ser un tema controvertido. Puesto que el tratamiento quirúrgico puede garantizar la curación en la mayoría de los casos, no parece adecuado el empleo de un tratamiento como la radioterapia ya que no existe suficiente experiencia para su uso en el DFSP.
La mayoría de los casos y series publicados corresponden a DFSP extirpados con márgenes ajustados o positivos donde emplean la radioterapia posteriormente92,154. Por todo ello, la radioterapia se reserva para casos verdaderamente inoperables, cuando se desestime el tratamiento con imatinib o para tratamiento paliativo.
PronósticoEl factor de mal pronóstico más reconocido en el DFSP es la extirpación quirúrgica inadecuada con márgenes de resección positivos para tumor o muy próximos al borde quirúrgico5,27,94. Este hecho se relaciona directamente con la posibilidad de recidiva tumoral y con la evolución del DFSP a formas histológicas tipo fibrosarcoma y más indiferenciadas.
Desde el punto de vista clínico, los DFSP de gran tamaño y aquellos localizados en la cabeza y en el cuello parecen estar relacionados con un peor pronóstico155.
Histológicamente, la presencia de áreas de fibrosarcoma25,59,156 en el DFSP se ha relacionado con una evolución más agresiva, así como el aumento del índice mitótico, una mayor densidad celular25 y la mutación de la p5359.
La afectación ganglionar por un DFSP es un hecho muy poco frecuente. Son muy pocos los casos aislados que se han publicado de DFSP con metástasis ganglionares. Según la revisión de Rutgers et al.24, en los 913 casos de DFSP encontraron solamente 11 con metástasis linfáticas regionales, lo que supone el 1%. Este hecho significó un drástico empeoramiento del pronóstico, puesto que la mayoría de los pacientes murieron en los 2 primeros años tras la aparición de la afectación ganglionar. Por otra parte, las metástasis ganglionares son hasta 3 veces menos frecuentes que las viscerales5. Por todo ello no está indicada la linfadenectomía rutinaria en el DFSP.
En general, en la mayoría de los casos de DFSP no complicados la aparición de metástasis viscerales es un acontecimiento excepcional que tradicionalmente oscila entre el 1 y el 5%157; sin embargo, estos datos se basan en series antiguas donde quizá no se trataban correctamente determinados casos lo que aumentaba el riesgo de metástasis157. Por otra parte, con la aparición de imatinib se han publicado recientemente muchos casos de DFSP metastásicos que no hubieran sido publicados si no es por haber sido tratados con este fármaco. De tal forma que parece muy complicado calcular correctamente la frecuencia de metástasis en el DFSP.
Sí que se conoce bien que la mayoría de los casos metastásicos acontecen en DFSP que han recidivado en varias ocasiones o en aquellos con zonas de fibrosarcoma en su histología24,25,59. La localización más frecuente de las metástasis en el DFSP es el pulmón151,153 que supone hasta el 75% de los casos metastásicos5.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.


