

La reciente publicación de las directrices (guidelines) S3 europeas sobre el tratamiento sistémico de la psoriasis1 y la revisión de las directrices para intervenciones biológicas sobre la psoriasis de la British Academy of Dermatology2, poco tiempo después de la aparición de las españolas de las que somos coautores3, justifican por su trascendencia una revisión crítica (sin discutir exhaustivamente posibles errores puntuales, de trascripción o incluso concepto, que pueden aparecer en cualquier publicación) y algunos comentarios que consideramos de posible interés general para los lectores de Actas Dermosifiliográficas y para todos los dermatólogos implicados en el tratamiento de los pacientes con psoriasis.
IntroducciónLa finalidad de unas directrices basadas en la evidencia científica es mejorar el cuidado de los pacientes, teniendo en cuenta los datos de eficacia, seguridad, efectividad, preferencias y satisfacción de los pacientes. Las directrices constituyen una guía para la elección del tratamiento y su monitorización con el fin de mejorar la calidad asistencial4. Aunque existen instrumentos específicos, como por ejemplo el Appraisal of Guidelines Research and Evaluation4, para evaluar la calidad de la elaboración de unas directrices, el éxito final de estas viene determinado por la satisfacción de los usuarios y la mejora en el estándar de la atención médica a los pacientes que pueda derivarse de su uso.
La redacción de unas directrices basadas en la evidencia representa la culminación de un proceso en el que se destila la evidencia científica disponible con respecto al tratamiento de una enfermedad, se ofrece un resumen crítico y se establecen unas recomendaciones basadas en la fuerza de la evidencia disponible. Este proceso se basa en una revisión sistemática de los estudios o ensayos clínicos originales, una evaluación de las revisiones sistemáticas y metaanálisis (basados en los anteriores) disponibles y el cotejo de las posibles publicaciones de síntesis o sinopsis, mediante el empleo de bases de datos tales como PubMed, EMBASE, Cochrane, DARE, etc.
No cabe duda de que las directrices basadas en la evidencia son imprescindibles para poner al alcance de los clínicos una revisión sumaria de los elementos de juicio necesarios para la prescripción de un tratamiento u otro, basada en una metodología rigurosa, pero no sustituyen en modo alguno al juicio clínico y la prescripción individualizada y anteponen el beneficio del paciente a toda otra consideración.
La base legal del tratamiento es, en cualquier caso, la ficha técnica del producto, pero el estándar de la práctica médica, con implicaciones medicolegales, puede estar basado en una directriz. Por otra parte, las directrices basadas en la evidencia científica frecuentemente constituyen un documento de referencia para la toma de decisiones con respecto a la financiación/reembolso del coste del tratamiento o a las limitaciones en la prescripción por parte de las autoridades sanitarias o las entidades que lo costeen, aunque por razones éticas siempre deberían primar, en último término, el beneficio individual del paciente y la libertad de prescripción aplicada a este.
Las directrices europeas y británicas recientemente publicadas presentan algunas limitaciones y deficiencias que consideramos oportuno detallar a continuación.
Representatividad (directrices europeas S3)Aunque existen grandes diferencias en los sistemas sanitarios y las prácticas de prescripción entre los diferentes estados europeos, e incluso en las diversas administraciones o divisiones territoriales dentro de un determinado Estado que justifican plenamente la publicación de directrices específicas, como las holandesas5, las británicas2,6, las alemanas7 o las españolas3, no cabe duda de que una directriz europea puede ser útil para facilitar el desarrollo de directrices individuales para cada país, e incorporar la experiencia de expertos a gran escala. Por este motivo resulta llamativo que en la elaboración de las directrices europeas —respaldada por la European Academy of Dermatology e iniciada hace unos años por el European Dermatology Forum, con un Guideline Committee de 23 miembros (uno solo español) y desarrolladas por un Subcommittee de 39 miembros – no fuera invitado a participar ningún experto español. Tampoco en la fase final de elaboración se tuvieron en cuenta las sugerencias de aquéllos a quienes gentilmente se nos ofreció el acceso al manuscrito poco antes de su publicación.
Consideraciones de eficaciaEn las directrices europeas sólo se contempla el tratamiento de inducción (10–16 semanas), lo que tiene sentido en tanto que representa el objetivo primario de los ensayos clínicos, base de la evidencia; en muchos ensayos, a partir de este momento, cesa el doble enmascaramiento, con lo que los pacientes que recibían placebo pasan a recibir el agente activo, y los otros continúan el tratamiento (fase de tratamiento «abierto»).
La diferente velocidad con la que alcanzan habitualmente su meseta de eficacia los diversos tratamientos puede ser importante cuando se requiere una instauración rápida del beneficio terapéutico, pero para muchos pacientes y clínicos el objetivo terapéutico debería evaluarse al cabo de aproximadamente 6 meses de tratamiento.
La variabilidad en cuanto al punto de corte en el que se evalúa el resultado primario del tratamiento tiene importantes implicaciones para los estudios de eficiencia8, al ser muy variables los intervalos de administración, que determinan el coste de cada fármaco por intervalo de tratamiento. Probablemente la unidad de tiempo ideal para evaluar la eficacia de un tratamiento sea el año, y deberían tenerse en cuenta tanto el peso del paciente como la respuesta y el carácter continuo o intermitente del tratamiento.
En las directrices debería incluirse, asimismo, siempre que fuera posible, la información disponible sobre el efecto a medio (24–26 semanas) y a largo (52–100 semanas) plazo, así como el efecto de la retirada del fármaco (tiempo hasta la recaída), el retratamiento y, cuando proceda, el posible ajuste de dosis para optimizar la respuesta terapéutica.
Aunque el porcentaje de pacientes que consiguen una mejoría igual o superior al 75% con respecto al valor basal del Psoriasis Area and Severity Index (PASI), o respuesta PASI 75, se considera el objetivo estándar de mejoría clínicamente significativa, tanto en la valoración de los ensayos clínicos9 como en la práctica clínica habitual10; para el paciente puede ser más importante la respuesta Physician Global Assessment de blanqueamiento o casi blanqueamiento (0–1) o conseguir una mejoría igual o superior al 90% con respecto al valor basal del PASI, o respuesta PASI 90; los correspondientes valores, cuando están disponibles, no siempre se recogen en las recomendaciones correspondientes a cada fármaco de las directrices publicadas.
La elección del «punto de corte» con respecto a la variable primaria de eficacia (porcentaje de pacientes que alcanza una respuesta PASI 75 a corto plazo) siempre es arbitraria (¿por qué el 60% y no el 50 o el 70%?); más importante como defecto metodológico es no tener en cuenta el efecto placebo en los ensayos clínicos; por este motivo, al igual que en los metanálisis11–13, debería enunciarse el riesgo relativo (RR) o la diferencia de riesgo con el placebo o el número de pacientes que hay que tratar para conseguir el resultado (number needed to treat en base 1 o 10), ya sea la respuesta PASI 75, PASI 90 o el objetivo terapéutico que se establezca.
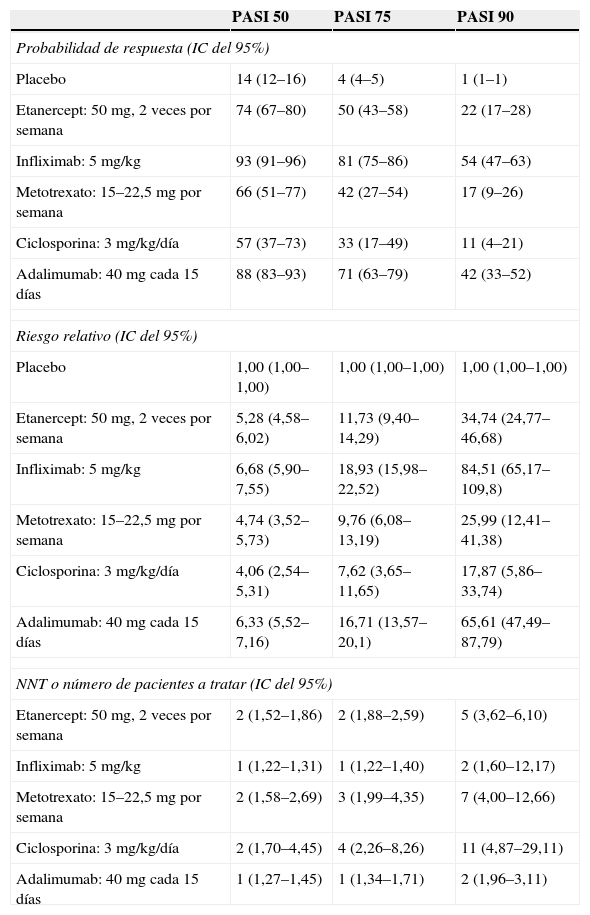
Para simplificar la representación de los resultados de eficacia (o, en su caso, de seguridad) en las directrices puede emplearse una escala cualitativa (por ejemplo, mediante cruces, flechas, etc.), pero en rigor deben presentarse los datos numéricos, y dejar el punto de corte al arbitrio del lector y del consiguiente prescriptor. Por ejemplo, a partir de los datos presentados en la tabla 1, el prescriptor puede elegir su punto de corte preferido basado en la probabilidad de respuesta (50, 70, 80, etc.), el RR (10, 15, 18, etc.) o el número de pacientes que hay que tratar (1, 2, etc.) para conseguir una respuesta PASI 75 (¿por qué no PASI 90?) que deje por encima uno, 2 o los 3 agentes antifactor de necrosis tumoral (anti-TNF) que lideran las correspondientes columnas, pero esto no implica que en un paciente dado cualquiera de los biológicos no pueda ser una alternativa válida, o que en un paciente determinado un fármaco con un valor inferior en el metanálisis no sea preferible a otros con un valor superior. La representación gráfica de los datos de RR o probabilidad de respuesta (fig. 1), obtenidos a partir de metanálisis13 o de los estudios fundamentales de un determinado fármaco14,15 para un objetivo específico de eficacia, resulta de gran ayuda para la percepción rápida de la eficacia relativa de los diferentes tratamientos.
Representación tabular de los resultados de un metanálisis que resume la evidencia científica disponible para algunos de los tratamientos sistémicos de la psoriasis
| PASI 50 | PASI 75 | PASI 90 | |
| Probabilidad de respuesta (IC del 95%) | |||
| Placebo | 14 (12–16) | 4 (4–5) | 1 (1–1) |
| Etanercept: 50mg, 2 veces por semana | 74 (67–80) | 50 (43–58) | 22 (17–28) |
| Infliximab: 5mg/kg | 93 (91–96) | 81 (75–86) | 54 (47–63) |
| Metotrexato: 15–22,5mg por semana | 66 (51–77) | 42 (27–54) | 17 (9–26) |
| Ciclosporina: 3mg/kg/día | 57 (37–73) | 33 (17–49) | 11 (4–21) |
| Adalimumab: 40mg cada 15 días | 88 (83–93) | 71 (63–79) | 42 (33–52) |
| Riesgo relativo (IC del 95%) | |||
| Placebo | 1,00 (1,00–1,00) | 1,00 (1,00–1,00) | 1,00 (1,00–1,00) |
| Etanercept: 50mg, 2 veces por semana | 5,28 (4,58–6,02) | 11,73 (9,40–14,29) | 34,74 (24,77–46,68) |
| Infliximab: 5mg/kg | 6,68 (5,90–7,55) | 18,93 (15,98–22,52) | 84,51 (65,17–109,8) |
| Metotrexato: 15–22,5mg por semana | 4,74 (3,52–5,73) | 9,76 (6,08–13,19) | 25,99 (12,41–41,38) |
| Ciclosporina: 3mg/kg/día | 4,06 (2,54–5,31) | 7,62 (3,65–11,65) | 17,87 (5,86–33,74) |
| Adalimumab: 40mg cada 15 días | 6,33 (5,52–7,16) | 16,71 (13,57–20,1) | 65,61 (47,49–87,79) |
| NNT o número de pacientes a tratar (IC del 95%) | |||
| Etanercept: 50mg, 2 veces por semana | 2 (1,52–1,86) | 2 (1,88–2,59) | 5 (3,62–6,10) |
| Infliximab: 5mg/kg | 1 (1,22–1,31) | 1 (1,22–1,40) | 2 (1,60–12,17) |
| Metotrexato: 15–22,5mg por semana | 2 (1,58–2,69) | 3 (1,99–4,35) | 7 (4,00–12,66) |
| Ciclosporina: 3mg/kg/día | 2 (1,70–4,45) | 4 (2,26–8,26) | 11 (4,87–29,11) |
| Adalimumab: 40mg cada 15 días | 1 (1,27–1,45) | 1 (1,34–1,71) | 2 (1,96–3,11) |
El lector puede elegir el punto de corte y el parámetro de eficacia que estime más adecuado como recomendación, aunque en unas determinadas directrices puede darse un valor orientativo como determinante de la intensidad de la recomendación (por ejemplo, probabilidad de conseguir una respuesta PASI 75 superior al 60% o superior al 20% para el PASI 90, o un riesgo relativo superior a 10 o 50, respectivamente, o un NNT inferior o igual a 2 para ambas medidas de eficacia, por ejemplo) si se alcanza un consenso entre los autores. Adaptada de Bansback et al13.
IC: intervalo de confianza; NNT: number needed to treat; PASI: Psoriasis Area and Severity Index.
 de conseguir una mejoría igual o superior al 75% con respecto al valor basal del Psoriasis Area and Severity Index (PASI) (o respuesta PASI 75) tras el tratamiento de inducción (10–16 semanas) para diversos tratamientos sistémicos y biológicos13–15 de la psoriasis en placas moderada-grave. Cualquier punto de «corte» en el área sombreada (probabilidad de conseguir una respuesta PASI 75 de entre el 50–75%) podría considerarse válido como determinante de recomendación terapéutica en unas directrices, aunque al disponer de fármacos cada vez más activos (en cuanto al porcentaje de pacientes que alcanza el objetivo terapéutico preestablecido, mejoría máxima en el PASI y velocidad de instauración del efecto terapéutico) aumenta el nivel de exigencia de los dermatólogos y los pacientes en cuanto a la eficacia esperada del tratamiento.")
Representación gráfica de las probabilidades (con intervalo de confianza del 95%) de conseguir una mejoría igual o superior al 75% con respecto al valor basal del Psoriasis Area and Severity Index (PASI) (o respuesta PASI 75) tras el tratamiento de inducción (10–16 semanas) para diversos tratamientos sistémicos y biológicos13–15 de la psoriasis en placas moderada-grave. Cualquier punto de «corte» en el área sombreada (probabilidad de conseguir una respuesta PASI 75 de entre el 50–75%) podría considerarse válido como determinante de recomendación terapéutica en unas directrices, aunque al disponer de fármacos cada vez más activos (en cuanto al porcentaje de pacientes que alcanza el objetivo terapéutico preestablecido, mejoría máxima en el PASI y velocidad de instauración del efecto terapéutico) aumenta el nivel de exigencia de los dermatólogos y los pacientes en cuanto a la eficacia esperada del tratamiento.
Las características basales de gravedad (PASI) de los pacientes no permiten establecer a priori la selección del biológico, aunque la velocidad esperada de instauración del efecto o el peso del paciente deben tenerse en cuenta con vistas a efectuar una decisión terapéutica individualizada (seleccionar el tratamiento con un inicio de acción más rápido, una mayor tasa de respuesta o una posología ajustada al peso).
Puesto que el acto de la prescripción implica un proceso de decisión basado en múltiples atributos del fármaco, el paciente o la enfermedad, es discutible la utilización de juicios de valor (recomendable, sugerido, etc.) al establecer un determinado orden o preferencia de la prescripción. La decisión siempre se efectúa de forma individualizada teniendo en cuenta no solo la eficacia de la intervención terapéutica en comparación con el placebo u otras intervenciones, sino también los posibles efectos adversos, la conveniencia del paciente o del método de administración, la presencia de características individuales del paciente (peso, artritis, enfermedades concomitantes, contraindicaciones o situaciones que obligan a tener precauciones especiales, falta o pérdida de respuesta, previsión de tratamiento intermitente por viajes, embarazo, intervenciones, etc.) o del brote de la enfermedad (rebote, extensión o carácter inflamatorio, que requieren una respuesta rápida). Estas situaciones especiales generalmente no se tienen en cuenta al seleccionar a los pacientes que se incluyen en los ensayos clínicos, y a menudo determinan el perfil de eficacia y seguridad de cada fármaco en cada paciente.
Consideraciones de seguridadResulta una práctica común en las directrices confundir las «contraindicaciones» con las «advertencias y precauciones» de la ficha técnica. Por ejemplo, no es recomendable administrar ningún agente bloqueador del TNF a pacientes con insuficiencia cardiaca grave (clases iii-iv de la New York Heart Association), pero no puede considerarse una contraindicación absoluta en el caso de etanercept. Tampoco lo es la gestación: aunque es recomendable suspender el tratamiento con anti-TNF en caso de embarazo, la categoría B de la Food and Drug Administration indica que los estudios en animales no han demostrado riesgos fetales, aunque no existan estudios adecuados o bien controlados en mujeres embarazadas, o que aunque los estudios en animales hayan mostrado efectos adversos, los llevados a cabo en humanos no los han demostrado (esta última alternativa es francamente poco probable, puesto que la demostración experimental de efectos adversos impediría la realización de estudios en seres humanos). En «el período crítico de las primeras 12 semanas»2 cabe considerar que el paso transplacentario de una molécula IgG es despreciable, por lo que la recomendación en el caso de los anticuerpos monoclonales debería ser suspender el tratamiento a partir del conocimiento de la situación de embarazo para evitar el paso transplacentario del anticuerpo monoclonal a partir del segundo trimestre de gestación16.
En cuanto a las recomendaciones para la prevención de la reactivación de la tuberculosis también puede considerarse discutible tanto la pauta (que evidentemente depende de la situación en cada país) como el intervalo de tratamiento previo al inicio del tratamiento biológico (2 meses2, con nivel de evidencia 4) cuando no existe todavía un acuerdo entre expertos en cuanto a este (generalmente se espera un mes, pero no hay datos en contra de reducir el tiempo de espera cuando la gravedad del caso lo requiera).
Consideraciones económicas (implícitas y explícitas) y recomendaciones terapéuticasLas directrices británicas2,6 han venido efectuando recomendaciones basadas no en la evidencia científica, sino en razonamientos farmacoeconómicos o estrictamente presupuestarios, cuyo ámbito quizás no corresponda propiamente a unas directrices. El médico, sin menoscabo de su responsabilidad social y de contención del gasto para conseguir un empleo eficiente de los recursos disponibles, tiene un compromiso ético con el paciente, haciendo de valedor de este para que pueda recibir el mejor tratamiento disponible en cada caso. Así, por ejemplo, consideramos inaceptable (y nos congratulamos de que se haya eliminado en la edición vigente de las directrices británicas) la arbitrariedad de requerir un tiempo de espera de 6 meses con enfermedad grave para que los pacientes fueran candidatos a recibir agentes biológicos6, o la selección de infliximab sólo cuando fuera necesario un inicio de acción rápido o para pacientes con formas de psoriasis pustulosa o eritrodérmica6 que no están incluidas en la ficha técnica, ni fueron criterios de inclusión en los correspondientes ensayos clínicos.
En los ensayos clínicos efectuados, la mayoría de los pacientes presentaba psoriasis moderada a grave (PASI >10–12) y no existe ninguna evidencia publicada de que los pacientes con psoriasis más grave (PASI >20) respondan mejor o peor que los otros, por lo que no hay razón alguna para limitar a esta subpoblación el tratamiento con infliximab (o cualquier otro biológico)17. El razonamiento implícito de que habría que reservar los biológicos con una mayor probabilidad de respuesta (basada en los ensayos clínicos) para los pacientes más graves o como tercera línea de tratamiento no se justifica por evidencia científica alguna.
En la edición del presente año de las directrices británicas2 se ha eliminado este sesgo y se hace alguna observación implícita sobre la falta de evidencia científica para aprobar, como hace el National Institute for Health and Clinical Excellence, el empleo de infliximab sólo en pacientes con PASI superior a 20; no se han demostrado diferencias significativas en cuanto a la respuesta a infliximab basada en el PASI basal (porcentaje de pacientes con respuesta PASI 75 frente a placebo: el 70,9 frente al 2,3 con PASI basal <20; el 76,0 frente al 1,3 con PASI basal ≥20) o la naturaleza y el número de los tratamientos previos18. Sin embargo, se continúan haciendo recomendaciones discutibles, como por ejemplo relegar ustekinumab a un papel de tratamiento de rescate en caso de falta de respuesta a agentes anti-TNF (¿cuántos?), para lo que no existe ni se aporta ninguna justificación (excepto la «falta» de experiencia de seguridad y la posible comodidad de ir a la zaga de la experiencia de otras especialidades). Aunque la reciente incorporación de ustekinumab haya permitido incluir a pacientes con experiencia previa de tratamiento biológico, eso no justifica relegarlo a un papel de tratamiento de rescate, ni tampoco la relativa falta de experiencia con respecto a la seguridad, pese a que los ensayos clínicos efectuados con ustekinumab en pacientes con psoriasis han incluido en el seguimiento a más pacientes/año que cualquier otro biológico. Los resultados de los ensayos más recientes, en los que se incluyen análisis de subpoblaciones de pacientes con exposición previa a varios sistémicos o biológicos, confirman la eficacia de ustekinumab en estos pacientes, aunque resulta algo inferior a la de los pacientes sin experiencia previa de tratamiento biológico.
En la actualidad no existen datos que permitan establecer cuál es la mejor pauta en caso de falta o pérdida de respuesta frente al tratamiento con un anti-TNF; sin duda deben tenerse en cuenta factores tales como el peso del paciente en los tratamientos en dosis fijas, o la inmunogenicidad y el aclaramiento de cada biológico, por lo que ante un fallo o pérdida de eficacia de un biológico anti-TNF la decisión de sustituirlo por otro agente del mismo grupo (¿cuál?) o cambiar a ustekinumab requiere mucha experiencia y juicio clínico, y siempre debe tomarse de forma individualizada, no de manera automática siguiendo la recomendación de una directriz.
Un defecto atribuible a las directrices, tanto europeas como británicas, es asumir de forma acrítica las limitaciones en cuanto a la prescripción (indicación de los biológicos como tratamiento de segunda línea) de las fichas técnicas de la EMEA; en las directrices británicas2 se especifica que la fuerza de la recomendación es D, y el nivel de evidencia es 3, mientras que en las europeas no se establece la evidencia científica de esta restricción.
Las restricciones en la indicación del tratamiento, con independencia de la gravedad de la enfermedad, son atribuibles exclusivamente a motivaciones de limitación de costes, puesto que no existe ningún dato en los criterios de inclusión o el análisis de los resultados de los ensayos clínicos presentados a las autoridades reguladoras estadounidenses y europeas que indique que sólo sean candidatos a tratamiento biológico «los pacientes con fracaso previo, intolerancia o contraindicación a tratamientos sistémicos, incluidos ciclosporina, metotrexato o PUVA». Sobre la base de la evidencia disponible, la Food and Drug Administration ha ido aprobando los diferentes agentes biológicos para el tratamiento de la psoriasis moderada a grave, sin las restricciones en cuanto al tratamiento previo que incluye la EMEA en su ficha técnica. Los clínicos preocupados por el bienestar del paciente, aunque deben seguir en su prescripción la ficha técnica por razones legales (la alternativa es el uso compasivo) y para que las respectivas agencias estatales subvencionen el tratamiento, no tienen por qué asumir como propia (aunque la acaten) la antedicha cortapisa, puesto que no se sustenta en ninguna evidencia científica.
La decisión terapéutica debería individualizarse siempre en el contexto de una medicina personalizada, y el intercambio de tratamientos (que implica que siempre deba escogerse el tratamiento con una mayor probabilidad de conseguir una determinada respuesta) es un planteamiento erróneo que asumen, en ocasiones, los responsables de la toma de decisiones alejados de la práctica clínica cotidiana. La respuesta individual de un paciente concreto en un momento determinado no puede predecirse a partir de los resultados de los ensayos clínicos en grupos de pacientes con características muy diferentes de las que se observan en la práctica clínica, y una respuesta PASI 75 puede ser excelente o completamente insuficiente para un paciente determinado, dependiendo de la situación.
Las consideraciones económicas o farmacoeconómicas, aunque deben especificarse en las directrices como un importante elemento de juicio, deberían ser ajenas a la decisión terapéutica en sí, y en todo caso determinarían posibles limitaciones con respecto al reembolso o no del tratamiento en cada sistema sanitario.
Las guías del National Institute for Health and Clinical Excellence19 no son más que unas recomendaciones para la prescripción con cargo al National Health Service en Inglaterra y Gales; constituyen un parámetro para justificar decisiones de reembolso en un contexto sanitario determinado, y no tienen en cuenta el máximo beneficio del paciente individual, sino la eficiencia, con vistas a la minimización de costes. Este último objetivo, aunque loable por cuanto permite acceder a más pacientes a un determinado tratamiento en un contexto de limitación de recursos, puede alterar la óptima calidad asistencial.
En general, en el caso de los tratamientos biológicos, la respuesta es mejor cuando la dosis depende del peso del paciente. Con el fin de aumentar la eficiencia del tratamiento pueden adoptarse políticas de reembolso adecuadas a cada medio sanitario para minimizar la diferencia de coste dependiente del peso20; otra alternativa sería pagar tan solo los tratamientos eficaces (se define eficacia, por ejemplo, como conseguir una respuesta PASI 75 o una respuesta Physician Global Assessment inferior o igual a 2 a las 12–16 semanas, que se mantiene posteriormente); las organizaciones de pacientes y los grupos de expertos de las academias nacionales de Dermatología deberían tener un importante papel (consultivo e incluso de participación en la toma de decisiones) en este sentido. Este tipo de medidas harían mucho más equitativa y eficiente la asignación de recursos, y permitirían una optimización del tratamiento basada no sólo en la evidencia científica (que aporta datos a corto plazo de poblaciones incluidas en ensayos clínicos), sino en la respuesta individual de cada paciente.
Conflicto de interesesLos autores han participado en ensayos clínicos, asesorías y conferencias con el patrocinio de Abbott, Janssen, MSD y Pfizer.


