


Mycosis fungoides is the most common cutaneous lymphoma. The probability of systemic involvement depends on disease extension and is very low during the early stages, when the clinical course is usually indolent. However, the risk of extracutaneous disease 20 years after diagnosis is 10% in patients with generalized plaques and 35.5% in patients with tumorous lesions.1 The most common finding is enlarged regional lymph nodes, although any organ can be affected, especially the lungs, spleen, liver, and gastrointestinal tract.2 Central nervous system (CNS) involvement is very uncommon, even more so in the absence of extracutaneous disease.
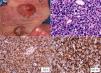
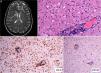
The patient was a 47-year-old woman with erythematous-desquamative lesions that first appeared 6 years previously and were diagnosed as stage 1b mycosis fungoides after 4 inconclusive biopsies. During a 4-year follow-up period she received treatment with potent topical corticosteroids, psoralen-UV-A, interferon, and oral bexarotene. During the last year, the patient developed tumorous lesions (Fig. 1A), with 14% Sézary cells (<1000/μL) in peripheral blood and unremarkable findings on a computed tomography (CT) scan of the chest, abdomen, and pelvis (stage IIb). The dose of oral bexarotene was increased and local radiation therapy started, and a partial response was observed. Skin biopsy of a rapidly growing tumorous lesion revealed large cell transformation with marked positivity for CD30 (Fig. 1B-D) that had not been present in the previous biopsies. A few months later the patient complained of recent memory loss and depression. The physical examination revealed nystagmus and dysmetria. An emergency CT scan of the brain ruled out acute disease. Similarly, a subsequent CT scan of the chest, abdomen, and pelvis revealed no significant abnormalities; nevertheless, the patient was admitted to hospital. Magnetic resonance imaging of the brain revealed diffuse white matter signal abnormalities, especially in the frontal region (Fig. 2A), which was compatible with infectious encephalitis. Analysis of cerebrospinal fluid revealed pleocytosis dominated by T lymphocytes with no atypia. Culture was negative. Biopsy of brain tissue revealed infiltration by CD4+CD8– T-cell lymphoma (Fig. 2B and C) with no large cell transformation and isolated CD30+ cells (Fig. 2D). Chemotherapy was started according to the BAM protocol (carmustine, methotrexate, and cytarabine). However, the disease progressed, with biopsy-confirmed kidney metastases and multiple pulmonary nodules compatible with metastasis. Large cell transformation was not observed in brain tissue or kidney tissue; therefore, the case was considered to be a localized phenomenon affecting the skin. Unfortunately, the patient died from respiratory insufficiency 3 months after the onset of neurological symptoms.
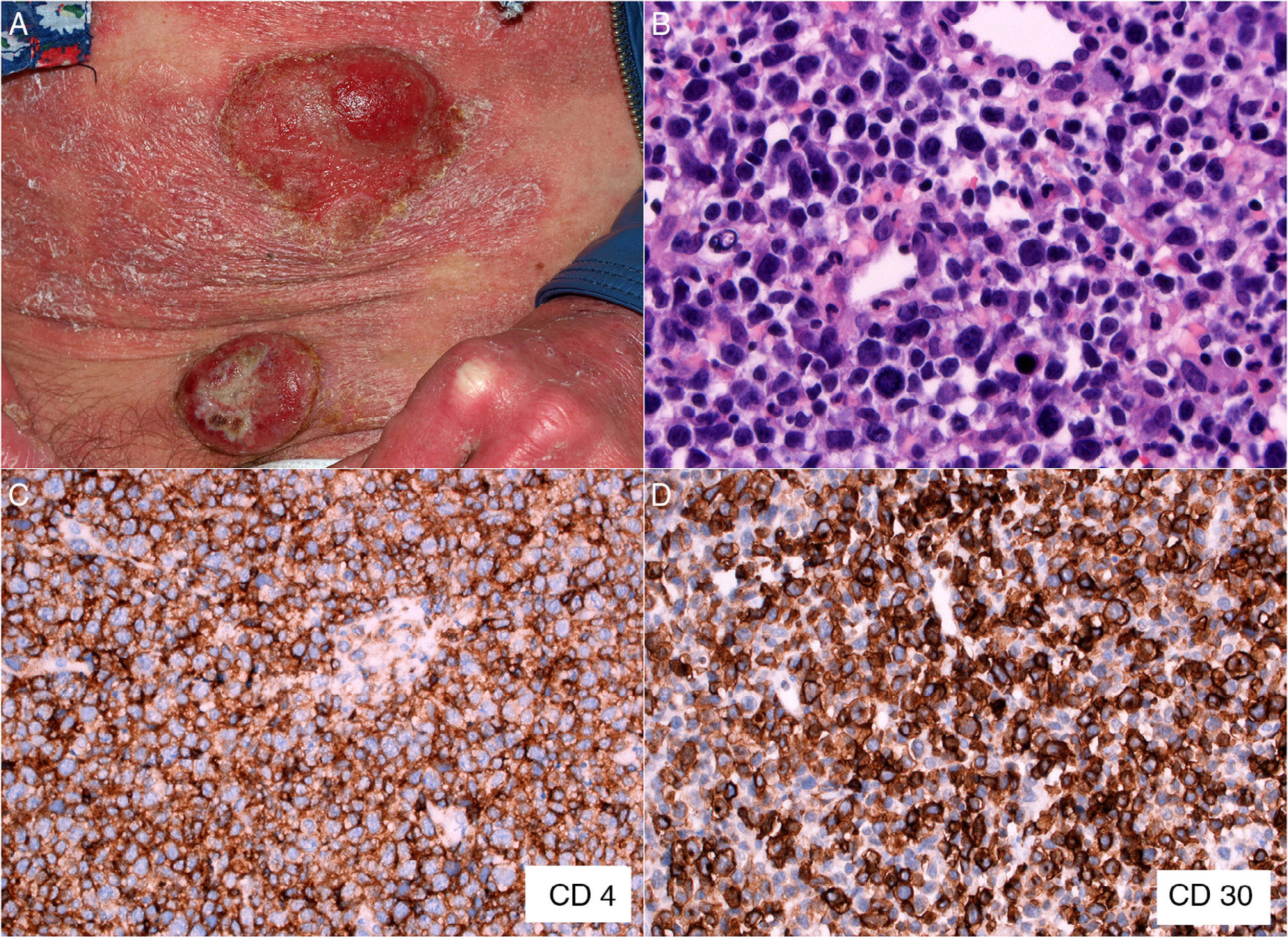
A, Mycosis fungoides in the tumor phase. B, Biopsy (detail) of a tumorous lesion. Note the diffuse large cell lymphoid infiltrate in the dermis (hematoxylin-eosin, original magnification, ×400). C, Immunohistochemical staining showing predominance of CD4+ T lymphocytes (CD4, ×400). D, Expression of CD30 antigen in cells of the lymphoid infiltrate (CD30, ×400).

A, Diffuse white matter signal abnormality, especially in the frontal and periventricular regions (arrows). B, Brain tissue biopsy showing a predominantly perivascular lymphoid infiltrate (hematoxylin-eosin, ×200). C, Immunohistochemical staining showing the CD4+ lymphocytic infiltrate in brain tissue (CD4, ×100). D, Expression of CD30 antigen in sparse lymphoid cells of brain tissue (CD30, ×40).
In contrast with other cutaneous lymphomas, such as aggressive epidermotropic CD8+ T-cell lymphoma, involvement of the CNS as a result of mycosis fungoides is exceptional, accounting for 1.6% of a series of 187 patients.3 However, CNS involvement was found in 11% to 14% of autopsies performed on patients who died of mycosis fungoides.4 Some authors report meningeal involvement to be the most common finding,4 whereas others claim that intraparenchymal brain involvement is the most common, with both forms able to occur simultaneously.5 The male-to-female ratio for this disease is 4:1, and in most cases, symptoms appear between 3 and 5 years after diagnosis of mycosis fungoides.5 The most frequent symptoms include confusion and depression, which are associated with lethargy and nausea. Optic and facial nerve involvement is common, as are gait disorders. The prognosis is poor, with a mean survival of 4.5 months after diagnosis.5
As for risk factors associated with CNS involvement, the series of Stein et al.6 showed that patients with at least 2 of 4 risk factors (T3-T4, N3, M1, and B1) had a 1 in 6 risk of CNS involvement at 10 years of follow-up compared with a 1 in a 100 risk in patients with 1 or none of the 4 risk factors.
The literature contains barely 10 cases of mycosis fungoides affecting the CNS associated with large cell transformation.5,7,8 Patients with mycosis fungoides and large cell transformation have a 5% probability of CNS involvement, compared with 1.6% of patients with no large cell transformation.8 Furthermore, while patients with large cell transformation usually present extracutaneous symptoms before cerebral involvement (lymph nodes and viscera), some patients with large cell transformation develop cerebral metastasis without previously having developed extracutaneous symptoms, as in the case we present.8 These patients should have an annual cerebral CT scan.8 As for CD30 expression, some studies show a better prognosis in patients with large cell transformation and CD30+ cells.9,10
Mycosis fungoides very rarely affects the CNS. Patients with large cell transformation are usually at increased risk, sometimes without previously having developed extracutaneous symptoms. Such patients should undergo radiological evaluation, and physicians should be on the alert for neurological symptoms.
Please cite this article as: de Quintana-Sancho A, Acebo-Mariñas E, Gardeazabal-García J, Aperribay-Esparza A. Micosis fungoide con afectación cerebral como primera manifestación extracutánea. Actas Dermosifiliogr. 2015;106:693–695.


