






El carcinoma de células de Merkel (CCM) es un tumor cutáneo muy agresivo y de mal pronóstico, aunque la incidencia es muy baja. Existen pocas series que analicen la experiencia en un mismo centro.
MetodologíaEstudio observacional, descriptivo y retrospectivo de todos los pacientes diagnosticados en un hospital de tercer nivel entre 2002 y 2017. Se recogieron las características epidemiológicas, clínicas, histológicas, el tratamiento y la supervivencia, y se dividió la muestra en 2períodos para el análisis (2002-2009 y 2010-2017). Se realizó un análisis de supervivencia mediante el modelo de Kaplan-Meier y un análisis multivariante mediante el modelo de riesgos proporcionales de Cox.
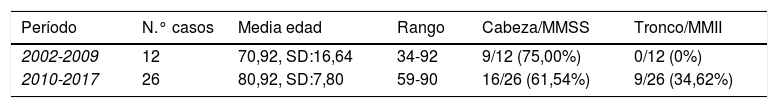
ResultadosSe incluyó a 38 pacientes, 24 hombres y 14 mujeres, con una edad media 77,76 años. El período medio de seguimiento fue de 30,11 meses. Se observó un aumento del 116% (12 vs. 26) entre los años 2002-2009 y 2010-2017, así como una edad media más avanzada (70,92 vs. 80,92; p<0,05) y un incremento de lesiones en tronco y miembros inferiores (34,62% vs. 0%). Once pacientes fallecieron debido al CCM. La supervivencia global a los 12 meses en la serie fue del 78,2% y a los 24 meses del 69,3%. Los factores asociados a mortalidad fueron la edad mayor de 70 años y la afectación ganglionar, mientras que la localización en miembros superiores y la realización de ampliación de márgenes aumentó la supervivencia. Al realizar el análisis multivariante, solo la afectación de ganglios permaneció como factor pronóstico.
ConclusionesSe ha observado un aumento de la frecuencia en los últimos años y un cambio en la forma de presentación a edades más avanzadas y en otras localizaciones diferentes a las clásicas.
Merkel cell carcinoma is a rare yet aggressive cutaneous tumor with a poor prognosis. Few studies have analyzed series of patients from the same hospital.
MethodologyWe performed a retrospective, descriptive, observational study of all patients diagnosed with Merkel cell carcinoma at a tertiary care hospital between 2002 and 2017. We recorded epidemiological, clinical, and histologic data and information on treatments and survival. For analysis, the sample was divided into 2 groups from different periods: 2002-2009 and 2010-2017. We performed survival analysis using Kaplan-Meier curves and multivariate analysis using a Cox proportional hazards model.
ResultsThirty-eight patients (24 men and 14 women) with a mean age of 77.76 years were included. Mean follow-up time was 30.11 months. On comparing 2010-2017 with 2002-2009, we observed a 116% increase in the number of Merkel cell carcinoma cases (26 vs. 12), an older mean age at diagnosis (80.92 vs. 70.92 years, P<.05), and an increase in lesions located on the trunk and lower limbs (0% vs. 34.62%). Eleven patients died of Merkel cell carcinoma. Overall survival was 78.2% at 12 months and 69.3% at 24 months. In the univariate analysis, age over 70 years and lymph node involvement were associated with mortality, while tumor location on the upper extremities and wide surgical excision were associated with improved survival. Only lymph node involvement retained its prognostic significance in the multivariate analysis.
ConclusionsIn this series, we observed that Merkel cell carcinoma has become more common in recent years and is now diagnosed at an older age and found in new anatomic locations.
El carcinoma de células de Merkel (CCM) o carcinoma neuroendocrino cutáneo primario es un tumor cutáneo muy agresivo y de mal pronóstico, con tendencia a la recidiva locorregional y metástasis a distancia, duplicando la mortalidad del melanoma1.
Afortunadamente, presenta una baja incidencia, por lo que existen pocas publicaciones que incluyan un número considerable de casos tratados en un mismo centro2-4.
Clínicamente, se caracteriza por ser tumores asintomáticos, de rápido crecimiento, predominio en áreas fotoexpuestas y presentarse con mayor frecuencia en pacientes ancianos e inmunodeprimidos, habiéndose desarrollado el acrónimo AEIOU (Asymptomatic; Expanding rapidly; Inmunosuppressed; Age>50; UV exposed) para recordar estas características5.
Estudios recientes reflejan un incremento de su incidencia en los últimos años, llegando incluso a triplicarse en algunas series, por una parte por el aumento de los factores de riesgos asociados conocidos (fundamentalmente, envejecimiento e inmunosupresión) y, por otra, probablemente por un mayor conocimiento de la enfermedad por parte de dermatólogos y patólogos1,6,7.
Para el diagnóstico es imprescindible la realización de un panel inmunohistoquímico. La detección de filamentos intermedios, principalmente CK20 y marcadores neuroendocrinos como CD56, cromogranina o sinaptofisina, y la ausencia de expresión de TTF1, MASH1, CK7, proteína S-100 y antígeno leucocitario común es útil para diferenciar el CCM de otros tumores de células redondas y azules de la piel8.
Los objetivos de este trabajo son conocer las características demográficas y clínicas de los pacientes con CCM de nuestra población, comprobar el aumento de los casos incidentes, así como analizar los factores pronósticos clínicos que influyen en la supervivencia.
Material y métodosEstudio observacional, descriptivo y retrospectivo de todos los pacientes diagnosticados de CCM en un hospital de tercer nivel en un período de 16 años (2002-2017) a través de los registros electrónicos de la historia digital y la base de datos de anatomía patológica. Todos los casos fueron confirmados histológicamente. Se recogieron las características epidemiológicas, clínicas (incluyendo el acrónimo AEIOU), histológicas y tratamiento empleado, recidiva así como la supervivencia a fecha 28 de febrero del 2018, incluyendo la causa de muerte en su caso. Todos los pacientes fueron tratados según práctica clínica habitual. La mayoría de los pacientes que, sin diagnóstico clínico, fueron diagnosticados histológicamente de CCM, fueron tratados mediante una segunda cirugía con ampliación de márgenes de 1 o 2cm tras el diagnóstico histológico. El estado general y la edad avanzada de algunos pacientes hicieron desestimar dicha ampliación. La biopsia selectiva del ganglio centinela (BSGC) no se realizó de manera sistemática, habiéndose realizado en pocos casos, por lo que esta variable no ha sido incluida en el análisis. En caso de positividad de la BSGC o la aparición de adenopatía, se realizó linfadenectomía. Los casos de enfermedad local avanzada irresecable fueron tratados mediante radioterapia locorregional, y en los casos de enfermedad diseminada con quimioterapia (regímenes basados en carboplatino y etopósido). En el último año del estudio, aquellos pacientes refractarios a tratamiento quimioterápico recibieron tratamiento con avelumab. Se calcularon las frecuencias absolutas y relativas (en porcentajes) para las variables cualitativas, y la media, la desviación estándar (SD), así como la mediana y el rango, para las variables cuantitativas. Para comparar las posibles diferencias en función del tiempo, se dividió la muestra en períodos de 8 años (2002-2009 y 2010-2017). Se realizó un análisis de supervivencia mediante el modelo de Kaplan-Meier y un análisis multivariante mediante el modelo de riesgos proporcionales de Cox. Todos los datos fueron recogidos y procesados mediante el paquete estadístico IBM Statistical Package for Social Sciences (SPSS), versión 21.0.0.0.
ResultadosPacientes. Se incluyó a 38 pacientes con CCM, cuyas características se resumen en la tabla 1. En total se encontró a 24 hombres y 14 mujeres (63,16% vs. 37,84%), con una relación 1,7:1, siendo el 100% de raza caucásica. La edad media al diagnóstico fue de 77,76 (SD: 12,07) años, sin diferencias entre sexos, con una mediana de 82 años (intervalo de 34 a 92 años). El período medio de seguimiento fue de 30,11 meses (SD: 36,83), con un rango entre 0 y 114 meses. Tan solo 3 (7,89%) pacientes presentaban algún tipo de inmunodepresión (síndrome linfoproliferativo, síndrome mieloproliferativo y trasplante cardiaco) y 9 (23,67%) antecedentes de neoplasias (4 hematológicas, 3 cáncer colorrectal y un caso de adenocarcinoma de glándula salival, próstata, vejiga y laringe, presentando un paciente 3neoplasias). Diez pacientes (26,32%) presentaban antecedentes personales de cáncer cutáneo no melanoma.
Características de los 38 pacientes con CCM
| Característica | Valor |
|---|---|
| Edad, media (rango) | 77,76 (34-92) |
| Sexo, n (%) | |
| Varones | 24 (63,16) |
| Mujeres | 14 (36,84) |
| Inmunosupresión, n (%) | |
| Sí | 3 (7,89) |
| No | 35 (92,11) |
| Antecedentes de neoplasias, n (%) | |
| Sí | 9 (7,89) |
| No | 21 (92,11) |
| Antecedentes CPNM, n (%) | |
| Sí | 10 (26,32) |
| No | 28 (73,68) |
| Sospecha inicial de Merkel, n (%) | |
| Sí | 6 (15,79) |
| No | 24 (84,21) |
| Sospecha inicial lesión maligna, n (%) | |
| Sí | 35 (92,11) |
| No | 3 (7,89) |
| Sospecha inicial, n (%) | |
| Espinocelular | 17 (44,74) |
| Basocelular | 10 (26,32) |
| Dermatofibrosarcoma | 3 (7,89) |
| Quiste | 3 (7,89) |
| Sarcoma partes blandas | 3 (7,89) |
| Queratoacantoma | 2 (5,26) |
| Estadio de presentación, n (%) | |
| Local | 30 (78,95) |
| Ganglionar | 8 (21,05) |
CPNM: cáncer de piel no melanoma.
En el momento del diagnóstico solo se sospechó CCM en 6 de los pacientes (15,79%). No obstante, en el 92% la sospecha clínica inicial fue de lesión maligna y solo un 8% se diagnosticó como lesión benigna inicialmente. El diagnóstico de sospecha más frecuente fue el carcinoma espinocelular (44,74%) seguido del carcinoma basocelular (26,32%), dermatofibrosarcoma protuberans, lesiones inflamatorias benignas y sarcoma de partes blandas (7,89%), y, por último, con el queratoacantoma (5,26%).
Características tumorales. En la mayoría de los casos, el CCM se ha presentado como una lesión tumoral única, con un tamaño medio de 3,89cm (SD: 2,90) mediana de 2,8cm y un rango comprendido entre 0,5 y 12cm, asintomática en el 90% de los casos, ulcerada (60,52%) y con un rápido crecimiento en menos de 3 meses (55,26%) (tabla 2).
Características tumorales
| Localización, n (%) | |
| Cabeza | 13 (34,21) |
| Miembros superiores | 13 (34,21) |
| Miembros inferiores | 6 (15,79) |
| Tronco | 3 (7,89) |
| Ganglionar | 2 (5,26) |
| Glúteos | 1 (2,63) |
| Tamaño, n (%) | |
| <1 | 5 (13,16) |
| 1-2 | 4 (10,53) |
| > 2 | 27 (71,05) |
| No especificado | 2 (5,26) |
| Tamaño, cm (n=36), media (rango) | 3,89 (0,5-12) |
| Ulceración, n (%) | |
| Presente | 23 (60,52) |
| Ausente | 15 (39,48) |
| Crecimiento, n (%) | |
| Rápido (< 3 meses) | 21 (55,26) |
| Lento (> 3 meses) | 17 (44,74) |
| Tratamiento, n (%) | |
| Cirugía solo | 18 (47,37) |
| Cirugía + RT | 10 (26,32) |
| Cirugía + RT + QT | 3 (7,89) |
| Cirugía + QT | 2 (5,26) |
| Cirugía + RT + QT +IT | 1 (2,63) |
| Cirugía + QT + IT | 1 (2,63) |
| RT + QT | 1 (2,63) |
| QT | 1 (2,63) |
| Ninguno | 1 (2,63) |
| Recidiva local (n=35), n (%) | |
| Sí | 10 (28,57) |
| No | 28 (71,43) |
| Recidiva local, meses, mediana (rango) | 5,5 (1-25) |
IT: inmunoterapia; QT: quimioterapia; RT: radioterapia.
Las localizaciones más frecuentes fueron la cabeza y los miembros superiores (34,21% en ambos casos), seguidos por los miembros inferiores (15,79%), el tronco (7,89%) y finalmente los glúteos (2,63%). En 2 pacientes no se identificó tumor primario cutáneo (5,26%). Dos tercios de las lesiones asentaban en áreas fotoexpuestas.
Inmunohistoquímicamente, las células de Merkel mostraron características epiteliales y neuroendocrinas con positividad para citoqueratina 20 (31/33; 93,94%), para cromogranina (25/31; 80,65%) y para sinaptofisina (19/25; 76,00%). Sin embargo, la citoqueratina 7 y sobre todo el TTF-1 han resultado negativas en la mayoría de los pacientes (16/21; 76,19%, y 20/22; 90,91%, respectivamente).
En el momento del diagnóstico, solo 8 pacientes (21,05%) presentaban afectación ganglionar, frente al 78,95% que presentaban solo afectación local. Ninguno de los casos presentó metástasis a distancia al inicio.
En cuanto al tratamiento, en la mayoría de los pacientes se empleó al inicio cirugía convencional (n=35) con posterior ampliación de los márgenes de entre 1 a 2cm en los casos en los que fue posible por la edad y localización (n=15), asociada a radioterapia locorregional (n=14) y a quimioterapia basada en esquemas de carboplatino y etopósido (n=7) en los casos de tumores o adenopatías no resecados en su totalidad, irresecables o en recaídas a lo largo de la enfermedad. Además, 2 pacientes han recibido tratamiento con avelumab, tras fracaso de la quimioterapia.
Análisis por períodos. Al dividir la muestra en 2períodos de años, 2002-2009 y 2010-2017, se observó un aumento del 116% de casos entre ambos períodos (12 vs. 26), así como unas características clínicas diferentes: por una parte, una edad media significativamente más avanzada (70,92 vs. 80,92; p<0,05) y por otra un cambio en la localización, ya que en el primer período todas las lesiones excepto 2 aparecieron en cabeza y miembros superiores (75%) y ninguna en tronco y miembros inferiores; sin embargo, en el segundo se observó un incremento de lesiones en tronco y miembros inferiores (34,62%), aunque sigue predominando la localización de cabeza y miembros superiores (61,54%) (tabla 3).
Distribución de casos y edad en los 2períodos del estudio
| Período | N.° casos | Media edad | Rango | Cabeza/MMSS | Tronco/MMII |
|---|---|---|---|---|---|
| 2002-2009 | 12 | 70,92, SD:16,64 | 34-92 | 9/12 (75,00%) | 0/12 (0%) |
| 2010-2017 | 26 | 80,92, SD:7,80 | 59-90 | 16/26 (61,54%) | 9/26 (34,62%) |
MMII: miembros inferiores; MMSSS: miembros superiores.
Supervivencia. A lo largo del período de seguimiento, recidivaron 10 de los 35 pacientes que se sometieron a cirugía (28,57%). La mediana de tiempo de recurrencia fue de 5,5 meses.
Al final del período de seguimiento, 17 (44,74%) pacientes habían fallecido, entre los cuales 11 (28,95%) pacientes debido al CCM y 6 (15,79%) por otras causas. La supervivencia específica en la serie fue del 78,2% a los 12 meses y del 69,3% a los 24 meses (fig. 1). Separando por estadios, la supervivencia específica fue del 87,0% a los 12 meses para estadios localizados al diagnóstico frente a 46,90% a los 12 meses para estadios ganglionares (fig. 2).


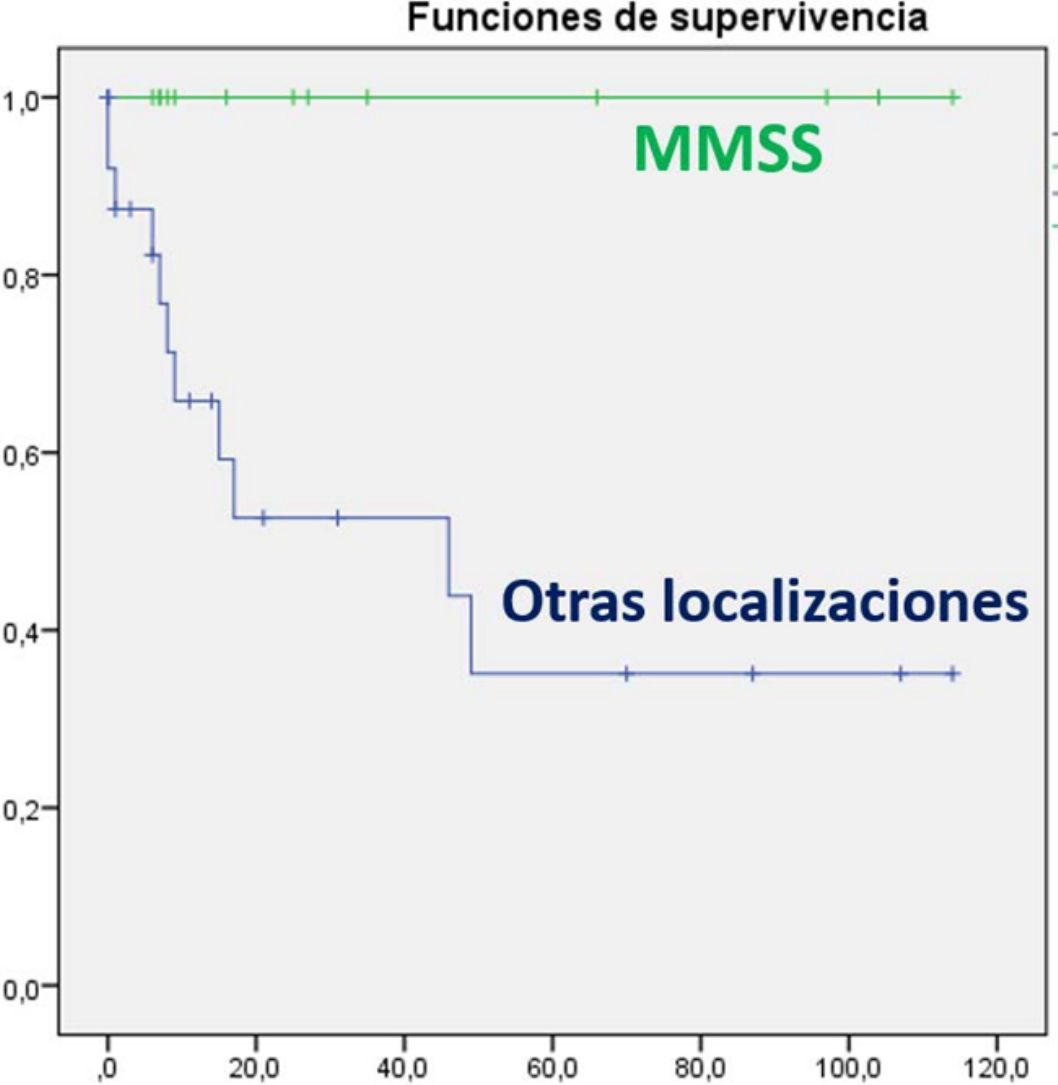
En el análisis univariante ha resultado estadísticamente significativa la edad al diagnóstico, asociándose los pacientes mayores de 70 años a mayor mortalidad (p=0,049). También se ha relacionado con mejor pronóstico la localización en miembros superiores (p=0,005) (fig. 3), presentando estos pacientes una supervivencia del 100% a pesar de tener mayor tamaño medio (4,64cm [SD: 3,98] miembros superiores vs. 3,60cm [SD: 2,41] otras localizaciones). Otros factores que se han relacionado con mayor supervivencia son la realización de ampliación de márgenes (para cualquier margen) tras la cirugía (p=0,011) y la ausencia de afectación ganglionar en cualquier momento de la enfermedad (p=0,023). Además se ha objetivado mayor supervivencia en el sexo femenino aunque sin alcanzar la significación estadística. Al realizar el análisis multivariante, mediante el modelo regresión de Cox, en el que se incluyeron estas 4variables significativas, solo la afectación de ganglios permaneció como factor pronóstico independiente de manera estadísticamente significativa.
Discusión
El CCM es un tumor muy poco frecuente, con pocas series de casos tratados en un mismo centro publicadas9,10. La serie más numerosa recoge a 9.387 pacientes procedentes de más de 1.000 hospitales de EE. UU.9. Las series españolas son escasas11,12, siendo la mayor la del Instituto Valenciano de Oncología, que recoge 48 casos presentados como póster en el 43.° Congreso Nacional del año 201513. En la serie que se presenta se recogieron un total de 38 casos en un período de 16 años, siendo por lo tanto una de las más numerosas.
Además de ser un tumor poco frecuente, tiene una apariencia inespecífica, incluso en ocasiones un aspecto benigno, por lo que la sospecha clínica inicial es poco habitual. Por ello, en 2008, Heath et al.5 crearon el acrónimo AEIOU con el fin de intentar resumir las características clínicas más importantes del CCM y de esta forma no retrasar el diagnóstico. A asintomático, E expansión rápida, I inmunodepresión, O(older) mayor de 50 años y U relacionado con exposición ultravioleta. La presencia de 3 o más de estas características son altamente sensibles para diagnosticar el tumor, ya que el 89% de los pacientes del grupo de Heath y en el 87% de pacientes de nuestra serie presentaban 3 o más criterios del AEIOU. Además del papel que desempeñan las radiaciones ultravioleta y la inmunodepresión en la patogenia, se ha descubierto en el año 2008 la implicación del poliomavirus, estando presente hasta en el 80% de los tumores14.
El perfil básico demográfico de la presente serie es concordante a lo descrito en la literatura, aunque destaca en nuestro estudio una mayor proporción de tumores en miembros superiores (34% vs. 24%)9 y una mayor cantidad de tumores ulcerados que lo que se describe en la literatura (61% vs. 10,6%)10. Los pacientes también han presentado estadios más localizados a los reflejados en otras series (79% vs. 65%)9. Es destacable que en el segundo período de años (2010-2017) ha aparecido un mayor número de tumores, al igual que lo descrito en la literatura con un aumento de la incidencia en los últimos años8,15,16, una mayor edad media de presentación y en localizaciones diferentes de cabeza y miembros superiores, muy escasas en el primer período. Este aumento de incidencia parece relacionarse con el aumento de la población de riesgo con una población más envejecida, inmunodeprimida y fotoexpuesta, y también con un mayor conocimiento del tumor por parte del dermatólogo y del patólogo, con el uso de nuevas técnicas inmunohistoquímicas6. La mayor edad media de presentación y el aumento de número de casos en los últimos años que se ha detectado en la presente serie no han sido descritos previamente en la literatura.
Los principales factores clínicos relacionados con buen pronóstico en el CCM son el sexo femenino, la localización en miembros superiores, tumor primario nodal sin lesión cutánea, tumores menores de 2cm y ausencia de afectación ganglionar y metastásica, así como ausencia de inmunodepresión entre otros8. Nuestros pacientes con tumores en miembros superiores también han tenido mejor pronóstico, a pesar de tener mayor tamaño tumoral. Esta asociación ya ha sido descrita previamente10. Aunque no se ha determinado por completo la causa de este mejor comportamiento, se ha visto que los tumores en esta localización se asocian a mayor presencia de poliomavirus respecto a otras localizaciones, hecho que también se ha relacionado con mayor supervivencia17. Por otra parte, han presentado mayor mortalidad los mayores de 70 años y aquellos pacientes con afectación ganglionar18. Aquellos pacientes que se intervinieron en un segundo tiempo para realizar ampliación de márgenes (entre 1 y 2cm) también tuvieron menor mortalidad.
Hasta hace poco, no disponíamos de tratamientos médicos específicos para el CCM, por lo que se empleaban regímenes de quimioterapia convencional que tenían una efectividad limitada. En los últimos años han apareciendo nuevos tratamientos inmunoterápicos como el pembrolizumab19, y más recientemente el avelumab20, ya aprobados para el tratamiento del CCM metastásico, que presentan tasas de respuesta completas o parciales de hasta el 56% de los casos. Dos pacientes con progresión ganglionar han recibido tratamiento con avelumab como segunda línea tras la quimioterapia clásica, aunque con un tiempo de seguimiento inferior a 6 meses, por lo que no es posible valorar la respuesta.
Para concluir presentamos una serie de 38 casos de CCM. Se ha observado un aumento de la frecuencia en los últimos años con un posible cambio de tendencia en la forma de presentación a edades más avanzadas y en otras localizaciones diferentes a las clásicas, hasta ahora no objetivada por ningún estudio español. La supervivencia de nuestros pacientes viene determinada por la edad, la localización, la ampliación de márgenes y la afectación ganglionar. Es importante cuando nos encontremos ante una lesión que tenga 3 o más criterios del AEIOU pensar en el CCM dentro del diagnóstico diferencial.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


