


El síndrome de Aicardi–Goutières (AGS) – leucoencefalopatía inflamatoria con calcificaciones ganglionares basales– es una interferonopatía de tipo I (activación del sistema de interferón [IFN]), que causa un incremento de la expresión de los genes regulados por IFN1. La expresión fenotípica del AGS son lesiones heterogéneas y cutáneas, que pueden ser clave para lograr un diagnóstico2.
Este es el caso de un varón de 19 años procedente de República Dominicana, con padres no consanguíneos, e historia de 18 años de paraparesia espástica y retraso en el desarrollo cognitivo y motor. Tras ser sometido a cirugía abdominal en su país en 2014, el paciente experimentó un episodio psicótico aislado con mutismo de etiología desconocida.
En abril de 2020, acudió con lesiones acrales que asemejaban sabañones, que desarrolló al llegar a España. El examen histopatológico reveló la presencia de necrosis e infiltrado linfohistiocítico perivascular denso con edema endotelial y focos trombóticos en los vasos dérmicos. No se observó depósito de mucina dérmica. No se realizó la prueba de inmunofluorescencia directa (DIF). El resto de las pruebas diagnósticas (incluyendo ANA y anticuerpos antifosfolípidos) fue normal, recibiendo el paciente el alta con sospecha de lesiones de tipo sabañones probablemente asociadas a infección asintomática por SARS-CoV-2.
En mayo de 2021, tras una infección del tracto respiratorio superior, fue hospitalizado por alucinaciones, comportamiento atípico (parálisis y periodos ocasionales de mutismo y perplejidad con episodios de movimientos anormales y estado maniforme), y un brote nuevo de lesiones cutáneas dolorosas. Entre las pruebas adicionales realizadas en dicho momento, la imagen de resonancia magnética reveló únicamente la presencia de calcificaciones ganglionares basales intracraneales de distribución lineal. La topografía de las arterias lenticulostriadas reveló una distribución bilateral y simétrica, sin cambios parenquemáticos ni otros hallazgos asociados, también observados en el TAC cerebral. Sin embargo, ninguno de dichos hallazgos ayudó a esclarecer la etiología de la pseudocatatonia. No se realizó análisis del LCR (líquido cefalorraquídeo).
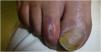
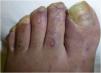
La dermatoscopia reveló la presencia de diversas placas bien delimitadas y deprimidas, de color porcelana de 3–6mm con un halo periférico eritematoso–violáceo en el dorso de los pies y dedos del paciente (figs. 1 y 2).


Dichas lesiones parecían similares a las descritas en la enfermedad de Degos (DD), que actualmente se considera una interferonopatía3. Ello, junto con las calcificaciones de los ganglios basales y los antecedentes clínicos de encefalopatía del paciente, fue indicativo de interferonopatía monogénica. Se solicitó un estudio de secuenciación del exoma completo, identificándose la variante homocigótica patogénica c.616C>T (p.His206Tyr) en el SAMHD1, lo cual respaldó el diagnóstico de AGS.
El AGS clásico es una enfermedad rara en la que se han identificado nueve genes implicados en el metabolismo intracelular del ácido nucleico con rasgos recesivos (mayoritariamente) o dominantes, y variantes patogénicas de pérdida (LOF) o ganancia (GOF) de función2. En dichas vías la desregulación desencadena una respuesta IFN-1 anormal, que es responsable de disfunción neurológica y de un rango amplio de fenotipos inflamatorios en otras enfermedades semejantes a AGS (STING, ADA2, DNAE2, LSM11, RNU7-1, DNASE1L3, ACP5, POLA1, USP18, OAS1, CDC42, STAT2, ATAD3A)1.
El AGS puede aparecer tras varios meses de desarrollo normal del niño, con síntomas inespecíficos tales como retraso psicomotor y/o pérdida de habilidades adquiridas, estabilizándose esta fase transcurridos pocos meses. Sin embargo, tras varios desencadenantes, pueden producirse brotes de signos extraneurológicos, principalmente cutáneos. De hecho, estos se presentan en hasta el 50% de la enfermedad relacionada con SAMHD1, que también tiene asociada vasculopatía intracerebral común2.
Las lesiones cutáneas que asemejan sabañones han sido descritas como lesiones dolorosas que se exacerban con el frío y se encuentran a menudo en las regiones acrales4. La gravedad de ellas varía desde acrocianosis y edema o placas eritematoso–violáceas a desarrollo de ampollas, ulceración, e incluso necrosis digital severa. Aunque se han reportado zonas de piel atrófica4, no hemos encontrado ninguna descripción similar a DD.
El examen histopatológico de los sabañones en los pacientes de AGS no arroja una visión uniforme. Puede presentarse un grado variable de vasculopatía trombótica, habiéndose reportado signos isquémicos secundarios tales como necrosis epidérmica, formación de bullas intraepidérmicas y cambios degenerativos como hialinización4.
Los síntomas psiquiátricos son excepcionalmente raros. De acuerdo con Varesio et al.5 en su serie de casos multicéntrica con 120 participantes de AGS, ningún paciente presentó síntomas psiquiátricos a lo largo del curso de la enfermedad. Actualmente, aún debe dilucidarse la asociación entre AGS y catatonia o pseudocatatonia, y los mecanismos subyacentes de síntomas psiquiátricos. Nosotros encontramos únicamente un caso similar al nuestro reportado por Ayrolles et al.6 en 2020. Ellos observaron altos niveles de IFN-α en el LCR seguidos de una rápida normalización tras la remisión clínica. Por tanto, la exacerbación de los síntomas psiquiátricos puede estar asociada a ciertos desencadenantes que pueden causar altos niveles de IFN-α, aunque la enfermedad estuviera previamente estabilizada.
Se ha mostrado el rol que juega IFN-I en el compromiso de los vasos sanguíneos tras el tratamiento de IFN-α con fenómeno de Raynaud, trombosis cutánea, y ulceración4. Además, las biopsias de las lesiones de DD han demostrado la firma de IFN-I en su patofisiología3.
En conclusión, las lesiones semejantes a Degos desencadenadas por el frío pueden ser una clave diagnóstica de interferonopatía genética. Por ello, el papel del equipo multidisciplinar que incluya a un dermatólogo en un paciente con signos cutáneos y psiquiátricos puede ser esencial para el diagnóstico temprano.
FinanciaciónEste estudio ha sido financiado por Instituto de Salud Carlos III a través del proyecto FIS-PI21/01642, y cofinanciado por la Unión Europea.
Conflicto de interesesLos autores declaran la ausencia de conflicto de intereses.


