

Sr. Director:
La esclerosis tuberosa, también llamada complejo de esclerosis tuberosa (CET o TSC, por sus siglas en inglés)1 es una enfermedad genética multisistémica, poco común, que causa tumores benignos en el cerebro y en otros órganos vitales como los riñones, el corazón, los ojos, el pulmón y la piel. Las manifestaciones renales son las que marcan el pronóstico de esta enfermedad, pues la aparición de angiolipomas renales origina hemorragias retroperitoneales e insuficiencia renal progresiva, principales causas de muerte en estos pacientes1.
La manifestación dermatológica más frecuente del TSC son los angiofibromas faciales (AF) múltiples bilaterales, hasta el punto de considerarse como un criterio mayor para establecer dicho diagnóstico1. No obstante, la presencia unilateral de estos AF es excepcional, y tan solo se han publicado 14 casos en la literatura (tabla 1)2–12. Otras manifestaciones cutáneas son las máculas hipomelanóticas, los hamartomas o nevos del tejido conectivo y los fibromas periungueales o subungueales. Presentamos un nuevo caso de AF múltiples de distribución unilateral sin otras manifestaciones de TSC.
Resumen de los casos descritos hasta la actualidad en la literatura de angiofibromas faciales múltiples unilaterales
| N.o Caso | Año publicado | Autor | Sexo | Edad | Edad inicio AF | Localización facial | Otros hallazgos |
| 1 | 1996 | McGrae2 | M | 23 | 5 | I | No |
| 2 | 1997 | Ankiller3 | H | 26 | 8 | I | No |
| 3 | 1998 | García Muret4 | M | 16 | 3 | D | Angiolipomas renales |
| 4 | 1998 | García Muret4 | H | 11 | 7-8 | D | No |
| 5 | 2000 | Silvestre5 | H | 5 | 2 | D | No |
| 6 | 2000 | Silvestre5 | H | 12 | 5 | I | Mácula hipopigmentada |
| 7 | 2002 | Del Pozo6 | M | 59 | 5 | I | No |
| 8 | 2003 | Trauner7 | H | 52 | 5 | I | Poliosis occipital |
| 9 | 2004 | Alonso8 | M | 39 | 20 | I | No |
| 10 | 2004 | Alonso8 | M | 37 | 9 | I | Amaurosis |
| 11 | 2004 | Sharma9 | M | 12 | 3 | I | Mácula hipopigmentada |
| 12 | 2004 | Mashhood10 | H | 7 | D | Fibromas periunguealesMácula hipopigmentadaCalcificaciones cranealesTumores retinianos | |
| 13 | 2006 | Camprubi11 | H | 13 | 3 | I | No |
| 14 | 2007 | Hall12 | M | 28 | 14 | D | No |
| 15 | - | Bordel | M | 40 | 15 | I | No |
AF: angiofibromas faciales; D: derecha; H: hombre; I: izquierda; M: mujer.
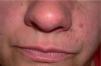
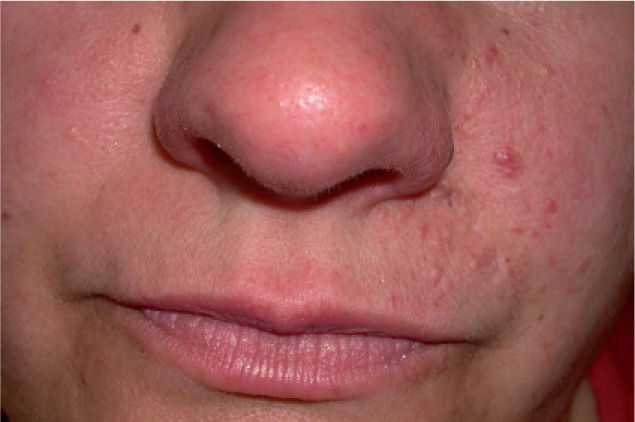
Se trata de una mujer de 40 años de edad, con antecedentes personales de catarata traumática en el ojo derecho, apendicectomía y en seguimiento periódico por el Servicio de Neurología por presentar una esclerosis múltiple en tratamiento con Inmunoferón®. Consultó por la aparición progresiva de pequeñas lesiones papulosas completamente asintomáticas, localizadas en el surco nasogeniano izquierdo, de crecimiento lento y de extensión progresiva desde los 15 años de edad. Dichas lesiones habían sido tratadas en varias ocasiones por su médico de Atención Primaria con queratolíticos, sin mejoría clínica aparente, lo que justificó su derivación a nuestra consulta. Carecía de antecedentes personales y familiares de cualquier enfermedad de tipo dermatológico, neurológico, renal o cardíaco, si bien refería que un hermano presentaba ocasionalmente crisis epilépticas no muy bien filiadas. No refería consanguinidad entre sus progenitores. La exploración física puso de manifiesto la presencia de múltiples pápulas cupuliformes de 2–4 mm de diámetro, del color de la piel normal, distribuidas aleatoriamente, pero concentradas en el surco nasogeniano y nasolabial izquierdo, sin sobrepasar la línea media (fig. 1). El resto de la exploración física, incluyendo una exploración neurológica exhaustiva, fue compatible con la normalidad. El examen con luz de Wood no evidenció la presencia de ninguna lesión hipopigmentada, ni tampoco observamos lesiones a nivel ungueal.
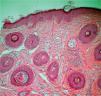
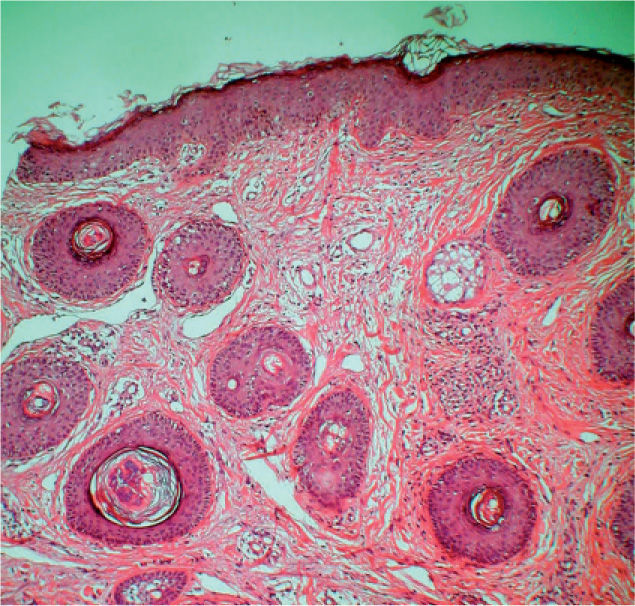
El estudio histopatológico de una de las lesiones cutáneas confirmó la sospecha clínica de AF, observando una fibrosis perivascular asociada con una hiperplasia angiomatosa (fig. 2). Se revisaron minuciosamente todas las pruebas de imagen realizadas por Neurología y, en vista de que la exploración física fue estrictamente normal, no se solicitó ninguna otra prueba complementaria. Aconsejamos el tratamiento con láser de CO2, que la paciente rechazó, y actualmente realiza controles periódicos.
El TSC es una enfermedad genética, de fenotipo muy variable, caracterizada por la tríada de retraso mental, epilepsia y AF faciales. Actualmente se considera un proceso hamartomatoso caracterizado por un trastorno de la proliferación, migración y diferenciación celular que se hereda con un rasgo autosómico dominante con una penetrancia variable, aunque el 60–70 % de los casos son esporádicos1. Estos casos esporádicos representarían mutaciones espontáneas nuevas, por ello carecen de historia familiar previa de dicha enfermedad como en nuestra paciente. Se produce por las mutaciones de los genes TSC1 y TSC2. Ambos genes determinantes TSC1 y TSC2 han sido identificados en los cromosomas 9q34 y 16p13.313.
Los AF múltiples bilaterales, antiguamente mal llamados adenomas sebáceos, son un criterio mayor del TSC, si bien se han encontrado en pacientes con neoplasia endocrina múltiple tipo 1 (NEM-I)14 y con neurofibromatosis tipo 115. Comienzan a desarrollarse en el área centro-facial durante los primeros años de vida (4–10 años) y llegan a afectar al 80–90 % de los pacientes con TSC7, manifestándose clínicamente como pápulas cupuliformes eritematosas, de superficie lisa y brillante. Están constituidos por tejido vascular y conectivo, y aunque son patognomónicos del TSC, su utilidad en el diagnóstico precoz es escasa, pues aparecen en la infancia tardía. La presencia unilateral de estos AF es excepcional y ha sido observada tan sólo en 14 casos en toda la literatura (tabla 1), 8 de los cuales carecían de otros criterios de TSC, grupo en el que añadimos a nuestra paciente. Sin embargo, los 6 pacientes restantes afectados de AF unilaterales presentaron otros hallazgos clínicos que apoyan el diagnóstico de TSC como máculas hipopigmentadas4,5,9,10, poliosis7, amaurosis8 o angiolipomas renales4.
El desarrollo unilateral de los AF ha mantenido un significado incierto a lo largo de los años. Los primeros casos descritos se atribuían a las diferentes formas de expresión del TSC, posteriormente se consideraron como uno de los primeros signos clínicos de aparición del TSC mientras que, en la actualidad, se consideran como una forma segmentaria, genéticamente bien definida del TSC4,5,8,10, producida por un mosaicismo genómico. La presencia de una mutación somática postcigótica tardía durante el período de desarrollo embrionario sería la responsable de este mosaicismo que nos explicaría la afectación de un solo segmento de la superficie corporal.
Los AF originan un aspecto antiestético de la cara, con ocasionales episodios de sangrado e incluso infecciones cutáneas (en lesiones vegetantes por la dificultad de aseo). Se han recomendado diferentes medidas terapéuticas, como resección, criocirugía, curetaje, dermoabrasión, láser de CO216, láser argón y láser diodo pulsado7,9,12. El láser de CO2 se ha utilizado con éxito en el tratamiento de estas lesiones, pero uno de los mayores problemas es la recidiva a largo plazo, ya que probablemente debido a la naturaleza de las lesiones éstas no se pueden eliminar permanentemente.
Es importante el diagnóstico precoz de estas formas segmentarias de TSC y aunque aún es una incógnita el tipo de exploraciones diagnósticas que debemos realizar y el consejo genético que deben recibir estos pacientes con AF unilaterales, al igual que otros autores, creemos imprescindible un seguimiento periódico de los mismos12. Este seguimiento es el que nos permitirá identificar de manera temprana la aparición de crecimientos tumorales u otras complicaciones, con la consecuente implantación de medidas terapéuticas oportunas.


