


Noonan syndrome with multiple lentigines (NSML, OMIM 151100) is a rare genodermatosis of autosomal dominant inheritance, high penetrance, and variable expressivity, previously known as the LEOPARD is an acronym for the major features of this disorder, including multiple Lentigines, ECG conduction abnormalities, Ocular hypertelorism, Pulmonic stenosis, Abnormal genitalia, Retardation of growth, and sensorineural Deafness.1 The skin signs play a key role in suspecting diagnosis and guiding genetic studies, as in the case described below.
This is the case of a 10-year-old male, child of non-consanguineous healthy parents, who was being monitored for short stature and learning difficulties. Physical examination revealed multiple 3mm to 4mm lentigines on the patient's face, neck, and trunk, with axillae and groin involvement (Fig. 1). Additionally, 6 brown macules with irregular borders were found distributed across the trunk, the largest measuring 3cm and being located on the patient's right buttock and (Fig. 2). A short neck and wide nasal bridge were also seen. Genetic testing revealed a heterozygous mutation c.1492C> T (R498W) in exon 13 of the PTPN11 gene (p. Arg498Trp). Cardiological and audiometric studies were normal.


In 85% of the cases, NSML is associated with mutations in the PTPN11 gene,2 which encodes the tyrosine phosphatase SHP2 protein involved in the RAS-MAPK signaling pathway involved in cell proliferation and differentiation processes. The pathogenesis of lentigenes development is explained by an increased melanin synthesis of melanocytes due to the SHP2 mutation, along with the activation of Akt/mTOR and STAT3 signaling pathways.3 The disruption of these signaling pathways involved in tumorigenesis can increase the risk of various neoplasms in NSML, including melanoma, with 5 cases being reported to this date.4
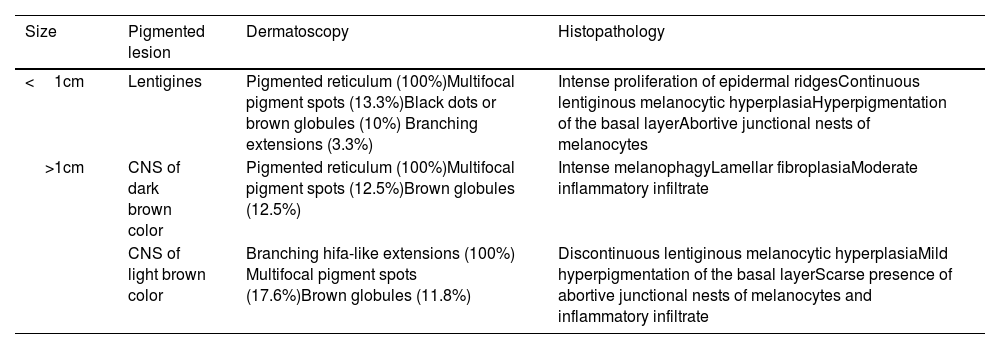
Skin signs are the most distinctive and common findings of this entity. Lentigines can be congenital but often appear around the age of 5 years increasing in number with age.5 They are dark brown lesions of a few millimeters in size, mainly located on the head and trunk without ever touching the mucous membranes. Cafe-au-lait spots are a common finding in nearly half of the patients and are darker in color than typical cafe-au-lait spots, hence the term “café noir spots.” Both the dermoscopic and histopathological characteristics of lentigines and café noir spots vary depending on their dark or light brown color (Table 1).6
Dermoscopic and histopathological features of lentigines and café noir spots (CNS) in NSML.
| Size | Pigmented lesion | Dermatoscopy | Histopathology |
|---|---|---|---|
| <1cm | Lentigines | Pigmented reticulum (100%)Multifocal pigment spots (13.3%)Black dots or brown globules (10%) Branching extensions (3.3%) | Intense proliferation of epidermal ridgesContinuous lentiginous melanocytic hyperplasiaHyperpigmentation of the basal layerAbortive junctional nests of melanocytes |
| >1cm | CNS of dark brown color | Pigmented reticulum (100%)Multifocal pigment spots (12.5%)Brown globules (12.5%) | Intense melanophagyLamellar fibroplasiaModerate inflammatory infiltrate |
| CNS of light brown color | Branching hifa-like extensions (100%) Multifocal pigment spots (17.6%)Brown globules (11.8%) | Discontinuous lentiginous melanocytic hyperplasiaMild hyperpigmentation of the basal layerScarse presence of abortive junctional nests of melanocytes and inflammatory infiltrate |
Affected individuals share phenotypic features with Noonan syndrome, such as facial dysmorphism (eyelid ptosis, hypertelorism, wide nasal bridge, and low-set ears), short stature, and pectus excavatum and carinatum.1,5 The most common and potentially life-threatening cardiac abnormality is hypertrophic cardiomyopathy, which occurs in up to 80% of all individuals.7 Less common are pulmonary stenosis and other mitral and aortic valve abnormalities. ECG conduction abnormalities are present in up to 75% of the cases. Among urogenital anomalies, bilateral cryptorchidism is notable in up to 50% of males, along with hypospadias, genital hypoplasia, and horseshoe kidney. Neurosensorial deafness affects 25% of the patients and, if present, intellectual disability is often mild.
Diagnosing NSML requires a high clinical suspicion, and genetic testing is often necessary for confirmation purposes. Differential diagnosis should include other rasopathies, especially Noonan syndrome and neurofibromatosis type 1. The phenotypic overlap with Noonan syndrome and the absence of lentigines in early childhood add complexity to its diagnosis.8
As far as we know, 12 missense mutations in the PTPN11 gene have been described for NSML,2 with limited reports on the R498W mutation to date, as seen in the case described above. While mutations in exon 13 of the PTPN11 gene have been associated with congenital heart disease in the NSML setting,9 this was not the case with our patient. Another recently published case shared a similar phenotype to the case presented here,10 stressing the lack of cardiac and auditory involvement. This highlights the broad phenotypic spectrum and variable expressivity of NSML, as well as the need to consider this condition even in the absence of hypertrophic cardiomyopathy or pulmonary stenosis. Additionally, within a family sharing the same mutation, there can be discordant phenotypes, especially regarding cardiac, genital, and skeletal anomalies.2
In conclusion, we should mention the role of dermatologists in the early diagnosis of NSML, since the initial clinical expression of the disease may be limited to the skin. When NSML is clinically suspected, genetic testing is advised, along with multidisciplinary and periodic assessments by cardiologists, endocrinologists, dermatologists, and other medical specialists.



