



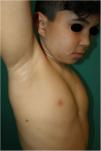
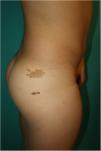

El síndrome de Noonan con múltiples lentigos (SNML OMIM 151100) es una genodermatosis infrecuente de herencia autosómica dominante con elevada penetrancia y expresividad variable, previamente conocida como síndrome LEOPARD, cuyo acrónimo recogía las características fenotípicas más frecuentes: lentigos, anomalías electrocardiográficas, hipertelorismo ocular, estenosis pulmonar, anomalías genitales, retraso del crecimiento y sordera neurosensorial1. Las manifestaciones cutáneas resultan clave para sospechar el diagnóstico y guiar el estudio genético como ocurrió en el caso que se describe.
Se trata de un varón de 10 años, hijo de padres sanos no consanguíneos, que hacía seguimiento por talla baja y dificultad para el aprendizaje. En la exploración física destacaba múltiples lentigos de 3-4mm en la cara, el cuello y el tronco, con afectación de axilas e ingles (fig. 1) Además, presentaba 6 máculas marrones de bordes irregulares distribuidas en el tronco, la mayor en la zona glútea derecha de 3cm (fig. 2). También se observaba un cuello corto y puente nasal ancho. En el estudio genético se demostró una mutación c.1492C>T (R498W) en heterocigosis (p. Arg498Trp) en el exón 13 en el gen PTPN11. El estudio cardiológico y la audiometría fueron normales.


En el 85% de casos, el SNML se asocia a mutaciones en el gen PTPN112, que codifica para la proteína tirosina-fosfatasa SHP2, que participa en la transducción de señales en la vía RAS-MAPK, implicada en procesos de proliferación y diferenciación celular. La patogénesis del desarrollo de los lentigos se explica por un aumento en la síntesis de melanina de los melanocitos, causado por la mutación en SHP2. A ello contribuye la activación de las vías de señalización de Akt/mTOR y STAT33. La alteración de estas vías de transducción, implicadas en la tumorogénesis, puede incrementar el riesgo de diversas neoplasias en el SNML, incluido el melanoma, habiéndose descrito cinco casos hasta la fecha4.
Las manifestaciones cutáneas son los hallazgos más distintivos y los más frecuentes en esta entidad. Los lentigos pueden ser congénitos, aunque suelen aparecer alrededor de los 5 años, aumentando en número con la edad5. Son lesiones de tonalidad marrón oscuro, de pocos milímetros que se localizan en el polo cefálico y el tronco, respetando las mucosas. Las manchas café con leche aparecen en aproximadamente la mitad de los pacientes y adquieren un tono más oscuro que las manchas café con leche típicas, por lo que reciben el nombre de máculas café negro. Las características dermatoscópicas e histológicas de los lentigos y las máculas café negro varían en función de su tonalidad marrón oscura o marrón clara (tabla 1)6.
Características dermatoscópicas e histológicas de lentigos y manchas café negro (MCN) del SNML
| Tamaño | Lesión pigmentada | Dermatoscopia | Histología |
|---|---|---|---|
| <1cm | Lentigo | Retículo pigmentado (100%)Manchas de pigmento multifocales (13,3%)Puntos negros o glóbulos marrones (10%)Extensiones ramificadas (3,3%) | Proliferación intensa de crestas epidérmicasHiperplasia melanocítica lentiginosa continuaHiperpigmentación de la capa basalNidos de melanocitos juncionales abortivosMelanofagia intensaFibroplasia lamelarInfiltrado inflamatorio moderado |
| >1cm | MCN tonalidad marrón oscuro | Retículo pigmentado (100%)Manchas de pigmento multifocales (12,5%)Glóbulos marrones (12,5%) | |
| MCN tonalidad marrón claro | Extensiones ramificadas tipo hifa (100%)Manchas de pigmento multifocales (17,6%)Glóbulos marrones (11,8%) | Hiperplasia melanocítica lentiginosa discontinuaHiperpigmentación de la capa basal leveEscasa presencia de nidos melanocitos juncionales abortivos y de infiltrado inflamatorio |
Los individuos afectos comparten rasgos fenotípicos con el síndrome de Noonan como la dismorfia facial, en forma de ptosis palpebral, hipertelorismo, puente nasal ancho y orejas de implantación baja. También pueden presentar talla baja y pectus excavatum y carinatum1,5. La anomalía cardíaca más frecuente y potencialmente letal es la miocardiopatía hipertrófica, que aparece hasta en el 80% de los individuos7. Menos frecuentes son la estenosis pulmonar y otras valvulopatías mitrales y aórticas. Los trastornos de conducción electrocardiográficos aparecen en hasta el 75% de los casos. Entre las anomalías urogenitales destaca la criptorquidia bilateral hasta en un 50% de los varones, así como hipospadias, hipoplasia genital y riñón en herradura. El 25% de los pacientes pueden desarrollar una sordera neurosensorial. La discapacidad intelectual, si está presente, suele ser leve.
El diagnóstico de SNML requiere una alta sospecha clínica, si bien suele ser necesario un análisis genético para su confirmación. El diagnóstico diferencial debe realizarse con otras rasopatías, en especial con el síndrome de Noonan y la neurofibromatosis tipo 1. El solapamiento fenotípico con el síndrome de Noonan y la ausencia de lentiginosis en los primeros años de vida añaden complejidad al diagnóstico8.
Hasta donde sabemos, se han descrito 12 mutaciones missense en el gen PTPN11 para el SNML2 y hay escasas publicaciones para la mutación R498W, que presentó el caso descrito. Si bien las mutaciones en el exón 13 del gen PTPN11 se han asociado a cardiopatía congénita en el contexto de SNML9, no fue así en nuestro paciente. Otro caso de reciente publicación10 compartía un fenotipo similar al del paciente presentado, ya que destacaba la ausencia de afectación cardiológica y auditiva. Ello pone de manifiesto el amplio espectro fenotípico y expresividad variable del SNML, así como la necesidad de considerar esta entidad, aun cuando no haya miocardiopatía hipertrófica o estenosis pulmonar. Incluso entre miembros de una familia que comparten la misma mutación puede haber fenotipos discordantes, especialmente en relación con las anomalías cardíacas, genitales y esqueléticas2.
En definitiva, es importante destacar el papel del dermatólogo en el diagnóstico precoz en el SNML, ya que la expresión clínica de la enfermedad puede verse al inicio limitada a la piel. Ante la sospecha clínica de SNML, se debe aconsejar un estudio genético, así como la evaluación multidisciplinar y periódica por parte de cardiólogos, endocrinólogos, dermatólogos y otros especialistas.


