






Las poroqueratosis son un grupo heterogéneo e infrecuente de dermatosis adquiridas o heredadas de etiología desconocida, caracterizadas por un trastorno de la queratinización secundario a una expansión clonal anormal de los queratinocitos. Se han descrito múltiples mutaciones genéticas potencialmente implicadas. Histológicamente, se caracterizan por la presencia de la lamela cornoide. Su presentación clínica es variable con formas localizadas, diseminadas e incluso eruptivas. Las poroqueratosis se han asociado con inmunosupresión, radiación ultravioleta, enfermedades sistémicas, infecciosas y neoplásicas. Muchos autores las consideran como entidades premalignas dada su potencial degeneración neoplásica a carcinoma escamoso o basocelular. Por ello, el seguimiento a largo plazo es uno de los pilares de su tratamiento, el que suele ser complejo y a menudo insatisfactorio. En la presente revisión se discuten los últimos avances en su etiopatogenia, diagnóstico y terapéutica, y se propone un algoritmo de tratamiento.
Porokeratosis comprises a group of heterogeneous and uncommon acquired or congenital skin diseases of unknown origin characterized by a keratinization disorder resulting from abnormal clonal expansion of keratinocytes. Numerous genetic mutations are thought to be involved. These conditions are characterized histologically by the presence of a cornoid lamella. Clinical manifestations are variable, with localized, disseminated, and even eruptive forms. Porokeratosis has been associated with immunosuppression, ultraviolet radiation, and systemic, infectious, and neoplastic diseases. Many authors consider it to be a premalignant condition because of the potential for malignant transformation to squamous cell or basal cell carcinoma. Therefore, long-term follow-up is a key component of treatment, which is usually complex and often unsatisfactory. We review the latest advances in our understanding of the pathogenesis, diagnosis, and treatment and propose a treatment algorithm.
Las poroqueratosis son un grupo muy infrecuente de dermatosis adquiridas o heredadas de etiología desconocida, caracterizadas por un trastorno de la queratinización. Se han descrito múltiples variantes clínicas, con unas características específicas de morfología, distribución y curso clínico. Muchos autores las consideran entidades premalignas dada la potencial degeneración neoplásica a un carcinoma escamoso o basocelular1,2. A continuación, se revisan diversos aspectos generales de las poroqueratosis, poniendo énfasis en las distintas formas clínicas: poroqueratosis actínica superficial diseminada (PASD), poroqueratosis superficial diseminada (PSD), poroqueratosis de Mibelli (PM), poroqueratosis lineal (PL), poroqueratosis diseminada eruptiva (PDE), poroqueratosis palmo-plantar y diseminada (PPPD) y poroqueratosis punctata (PP), así como algunas variantes descritas recientemente. Además, se resume la evidencia disponible de las opciones terapéuticas.
EpidemiologíaSon trastornos muy infrecuentes de los que se desconoce su incidencia y prevalencia exacta. En general, afectan a adultos. Sin embargo, algunas formas se pueden iniciar en la infancia, como la PM y la PSD1,2. No se han descrito diferencias étnicas o raciales1. Algunas series sugieren que predominan en hombres3,4, en especial la PM y la PPPD. Sin embargo, la PL y PASD serían más frecuentes en mujeres1. La PASD tiene una relación directa con la radiación ultravioleta (RUV), afecta principalmente a caucásicos y, consecuentemente, se observa una alta prevalencia en países como Australia1.
EtiopatogeniaSi bien se cree que las poroqueratosis se deben a una expansión focal clonal anormal de los queratinocitos, su etiopatogenia es desconocida2. La paraqueratosis podría ser secundaria a una maduración defectuosa de queratinocitos o a una epidermopoyesis acelerada. Se ha evidenciado una apoptosis prematura de los queratinocitos de la lamela cornoide, con una ausencia de la capa granulosa y una expresión defectuosa de loricrina y filagrina5.
Se han descrito múltiples factores involucrados:
- a.
Factores genéticos
Existe un número importante de casos familiares, con un patrón de herencia autosómico dominante de penetrancia variable. Esto ha sido descrito en PM, PL, PASD, PSD y PPPD1,2. Algunos casos esporádicos se asume que son debido a mutaciones somáticas. Se ha sugerido que las diversas formas de poroqueratosis serían fenotipos diferentes de una alteración genética común2. Se han descrito múltiples loci genéticos, siendo la PASD una de las más estudiadas, con cinco loci identificados en los cromosomas 1, 12, 15, 16 y 18; algunos cerca del gen de la enfermedad de Darier (12q23.2-24.1) y otros en un mismo locus de susceptibilidad a psoriasis (18p11)6-10. Un sexto loci ubicado en el cromosoma 12 ha sido descrito en la PPPD11, sin embargo, algunos autores creen que estos casos en realidad corresponderían a PM12.
Se ha relacionado la mutación de genes de la vía del mevalonato con la PASD, puesto que tiene un rol en la diferenciación queratinocitaria y en la protección de la apoptosis inducida por RUV13. La alteración de esta vía se ha identificado en el 33% de los casos familiares y en el 16% de los casos esporádicos con PASD14. Otros genes involucrados en la diferenciación epidérmica potencialmente implicados son: SSH1, SART3 y SLC17A915. Formas localizadas como la PM y PL pueden ser secundarias a un mosaicismo, por una pérdida focal de la heterocigosidad a partir de mutaciones somáticas. Esto explicaría la asociación de formas localizadas y PASD en algunos pacientes16.
- b.
Radiación ultravioleta
La RUV ha sido relacionada principalmente con la PASD a partir de estudios experimentales y observaciones clínicas que muestran la presencia de las lesiones cutáneas en las áreas fotoexpuestas, una exacerbación estival, una mayor incidencia en áreas geográficas con una exposición solar alta y por casos inducidos por la fototerapia1,17. En contrapartida, hay casos que mejoran con la fototerapia y la PASD no suele afectar el rostro1.
- c.
Inmunosupresión
Se han descrito casos asociados con una inmunosupresión, en especial en los trasplantados de órganos sólidos y de médula ósea1,18, a neoplasias hematológicas, VIH, fármacos, y enfermedades inflamatorias y/o autoinmunitarias. En los trasplantados renales se ha descrito una incidencia de poroqueratosis de hasta un 10%, con un período de latencia entre el trasplante y el inicio de la enfermedad de cuatro meses a 14 años (promedio de 4-5 años)19. Las formas clínicas más frecuentes en inmunosuprimidos son la PASD y PM, o un patrón mixto entre ambas variantes18.
El mecanismo que vincula la inmunosupresión y el desarrollo de una poroqueratosis es incierto. Algunos estudios muestran un aumento de la expresión de antígenos HLA-DR en las células de Langerhans epidérmicas en las lesiones de poroqueratosis19. Esto podría indicar una alteración de la inmunovigilancia que permitiría la proliferación clonal de unos queratinocitos anormales.
- d.
Otros
Se han descrito casos aislados asociados con fármacos (suramina, hidroclorotiazida, furosemida, hidroxiurea, gentamicina, exemestano y flucloxacilina)20,21, incluidos algunos agentes biológicos como etanercept, certolizumab y trastuzumab21-23.
Existen asociaciones con infecciones (virus del papiloma humano, virus herpes simple, virus hepatitis C [VHC] y leishmaniasis)1,24 y con algunas enfermedades sistémicas como la enfermedad de Crohn, la hepatopatía crónica, la insuficiencia renal crónica, la amiloidosis cardíaca primaria, la pancreatitis aguda, el síndrome de Sjögren, la artritis reumatoide, la miastenia gravis, la espondilitis anquilosante y la diabetes mellitus, esta última asociada principalmente con la PSD25-27. Las poroqueratosis también se han relacionado con algunas genopatías: la trisomía 16, el síndrome CAM (Craniosynostosis–anal anomalies–porokeratosis syndrome), el síndrome de rotura de Nijmegen, la fibrosis quística, el síndrome de Werner, la protoporfiria eritropoyética, el síndrome de Rothmund-Thomson y el síndrome de Bardet-Biedl1,28. Es posible encontrar asociaciones con otras dermatosis como el liquen plano, la psoriasis, los pénfigos, la hidradenitis supurativa, la alopecia areata, el pioderma gangrenoso, el lupus discoide, el vitíligo y el liquen escleroso1,29.
Una asociación de gran relevancia es con las neoplasias hematológicas y de órganos sólidos, descritas fundamentalmente en las poroqueratosis diseminadas eruptivas (PDE) (hasta en un tercio de los casos de PDE, muchos de ellos con criterios para considerarla una dermatosis paraneoplásica)30,31. Se ha sugerido que esta relación se debería a una inmunosupresión paraneoplásica, además de la coexistencia de infecciones virales como el VHC, relacionado con la inducción de mutaciones del supresor tumoral p5330.
Algunas descripciones de casos también han vinculado el desarrollo de una poroqueratosis con factores traumáticos como en el acceso vascular para hemodiálisis o quemaduras32,33. La electronterapia y la radiación ionizante también se han sugerido como desencadenantes34.
Poroqueratosis y transformación malignaLos queratinocitos de la lamela cornoide pueden exhibir diversos grados de displasia2,35. El mecanismo inicial para la malignización sería una pérdida alélica asociada con una sobre-expresión de p5336,37. Se ha sugerido la participación de otras proteínas, tales como la psi-3, la citoqueratina, la filagrina y la involucrina. La RUV puede actuar como un desencadenante al inducir mutaciones de p532.
La poroqueratosis se considera una alteración premaligna, ya que todas sus variantes tienen un potencial de malignización (más frecuentemente a un carcinoma escamoso y, en segundo lugar, a un carcinoma basocelular)35. Las lesiones malignas suelen ser únicas en dos de cada tres casos y se ubican frecuentemente en la piel no expuesta al sol de las extremidades, con un período de latencia de hasta 36 años2. La incidencia de malignización sería del 7,5-11%38,39 (tabla 1). El riesgo aumenta en las lesiones de mayor tamaño, pues se presentan con una epidermis hipertrófica y más alteraciones en la ploidía del ADN40.
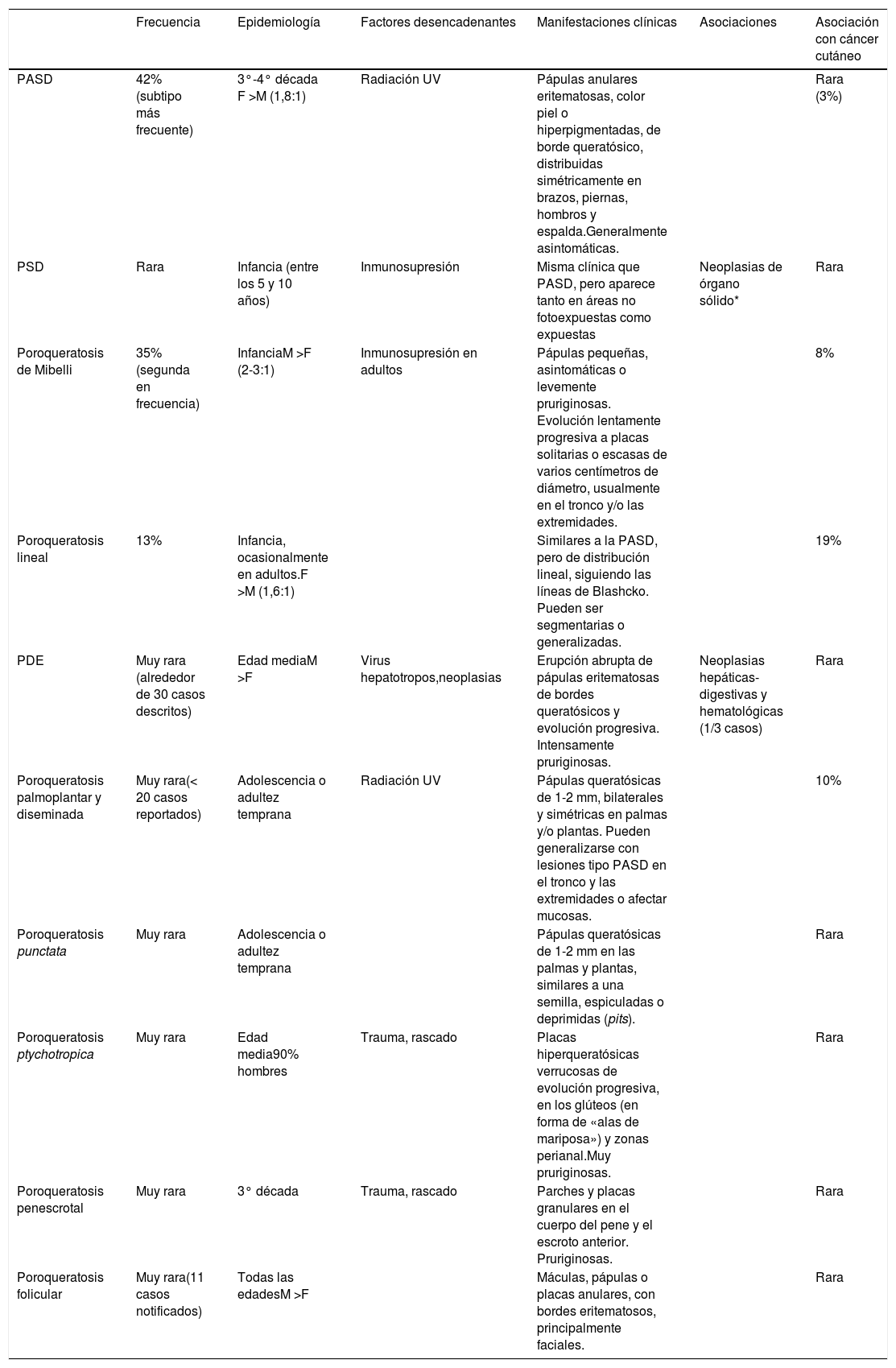
Características clínicas y epidemiológicas de los distintos subtipos de poroqueratosis
| Frecuencia | Epidemiología | Factores desencadenantes | Manifestaciones clínicas | Asociaciones | Asociación con cáncer cutáneo | |
|---|---|---|---|---|---|---|
| PASD | 42% (subtipo más frecuente) | 3°-4° década F >M (1,8:1) | Radiación UV | Pápulas anulares eritematosas, color piel o hiperpigmentadas, de borde queratósico, distribuidas simétricamente en brazos, piernas, hombros y espalda.Generalmente asintomáticas. | Rara (3%) | |
| PSD | Rara | Infancia (entre los 5 y 10 años) | Inmunosupresión | Misma clínica que PASD, pero aparece tanto en áreas no fotoexpuestas como expuestas | Neoplasias de órgano sólido* | Rara |
| Poroqueratosis de Mibelli | 35% (segunda en frecuencia) | InfanciaM >F (2-3:1) | Inmunosupresión en adultos | Pápulas pequeñas, asintomáticas o levemente pruriginosas. Evolución lentamente progresiva a placas solitarias o escasas de varios centímetros de diámetro, usualmente en el tronco y/o las extremidades. | 8% | |
| Poroqueratosis lineal | 13% | Infancia, ocasionalmente en adultos.F >M (1,6:1) | Similares a la PASD, pero de distribución lineal, siguiendo las líneas de Blashcko. Pueden ser segmentarias o generalizadas. | 19% | ||
| PDE | Muy rara (alrededor de 30 casos descritos) | Edad mediaM >F | Virus hepatotropos,neoplasias | Erupción abrupta de pápulas eritematosas de bordes queratósicos y evolución progresiva. Intensamente pruriginosas. | Neoplasias hepáticas-digestivas y hematológicas (1/3 casos) | Rara |
| Poroqueratosis palmoplantar y diseminada | Muy rara(< 20 casos reportados) | Adolescencia o adultez temprana | Radiación UV | Pápulas queratósicas de 1-2 mm, bilaterales y simétricas en palmas y/o plantas. Pueden generalizarse con lesiones tipo PASD en el tronco y las extremidades o afectar mucosas. | 10% | |
| Poroqueratosis punctata | Muy rara | Adolescencia o adultez temprana | Pápulas queratósicas de 1-2 mm en las palmas y plantas, similares a una semilla, espiculadas o deprimidas (pits). | Rara | ||
| Poroqueratosis ptychotropica | Muy rara | Edad media90% hombres | Trauma, rascado | Placas hiperqueratósicas verrucosas de evolución progresiva, en los glúteos (en forma de «alas de mariposa») y zonas perianal.Muy pruriginosas. | Rara | |
| Poroqueratosis penescrotal | Muy rara | 3° década | Trauma, rascado | Parches y placas granulares en el cuerpo del pene y el escroto anterior. Pruriginosas. | Rara | |
| Poroqueratosis folicular | Muy rara(11 casos notificados) | Todas las edadesM >F | Máculas, pápulas o placas anulares, con bordes eritematosos, principalmente faciales. | Rara |
PSD, poroqueratosis superficial diseminada; PASD, poroqueratosis actínica superficial diseminada; PDE poroqueratosis diseminada eruptiva; F, femenino; M, masculino; UV, ultravioleta.
Las poroqueratosis se manifiestan como unas pápulas o placas eritematomarronáceas bien delimitadas, de crecimiento centrífugo, y de tamaño y forma variables. Su borde está formado por una lámina queratósica fina, que apunta hacia el centro de la lesión, el cual puede ser deprimido o ligeramente atrófico y, con menor frecuencia, hiperqueratósico e hiperpigmentado1,2.
De acuerdo con el número, el tamaño, la forma y la distribución de las lesiones, se han descrito las siguientes variantes (tabla 1):
- a.
Poroqueratosis actínica superficial diseminada
La PASD es la forma más común de poroqueratosis (56% de todas las variantes clínicas)4 y se relaciona directamente con la fotoexposición. Se inicia generalmente en la tercera y cuarta década de la vida, con un predominio en mujeres (1,8:1). Se manifiesta con múltiples pápulas anulares pequeñas, que pueden llegar a ser cientos, y pueden confluir en placas policíclicas. Se distribuyen de forma simétrica en la espalda y la cara extensora de las extremidades1,2 (fig. 1A-B). Curiosamente, afecta al rostro sólo en el 15% de los casos41. Suelen ser asintomáticas, pero pueden ser pruriginosas hasta en un tercio de los pacientes1. Más de la mitad de los pacientes afectados refieren exacerbaciones durante el verano o tras la fototerapia42.
- b.
Poroqueratosis superficial diseminada

Es una forma similar a la PASD, pero en la que la RUV no puede considerarse como un desencadenante. Tiene un inicio precoz, entre los 5 y 10 años1,2. Existen casos de PSD coexistiendo con PL congénita en un lactante y un preescolar43,44.
Las lesiones se distribuyen en la piel, tanto fotoexpuesta como no fotoexpuesta, principalmente del tronco, los genitales y las regiones acrales (fig 1C-D), y puede afectar la boca2,45.
- c.
Poroqueratosis de Mibelli
Es la segunda forma clínica más frecuente3. Suele presentarse desde la infancia o adolescencia, aunque puede iniciarse en adultos, describiéndose un predominio en hombres (2-3:1)46. Se manifiesta con una o varias placas anulares con un centro atrófico o, con menos frecuencia, hiperqueratósico, de afectación unilateral y de crecimiento progresivo. El subtipo gigante puede superar los 20 cm y tiene un potencial de malignización alto1,47 (fig. 3A). Se distribuye generalmente en el tronco o las extremidades, aunque ha sido descrito el compromiso acral, facial, genital, del cuero cabelludo y de la mucosa oral1,2. Se han comunicado casos aislados con una posible variante hiperqueratósica, en la que las lesiones son verrucosas, múltiples y afectan a las extremidades48.
- d.
Poroqueratosis lineal

A. Poroqueratosis de Mibelli. Placa redondeada eritematosa de 4 cm con borde elevado e hiperqueratósico en la rodilla izquierda. El estudio histológico confirmó el diagnóstico. B. Poroqueratosis ptychotropica. Múltiples placas eritematodescamativas confluyentes de borde hiperqueratósico en zona perianal y glúteo derecho.
Es una forma infrecuente de poroqueratosis de predominio femenino (1,63:1)2. Para algunos autores no representaría una forma distinta de poroqueratosis, sino más bien una PM o PASD con una distribución lineal49. Se caracteriza por la presencia en las extremidades de múltiples pápulas o placas hiperqueratósicas agrupadas linealmente (fig. 2A-B). Suelen seguir un patrón Blaschkoide, aunque excepcionalmente pueden presentarse de forma segmentaria o generalizada, como una manifestación de mosaicismo genético50. Suelen presentarse de forma congénita, sin embargo, existen casos con un inicio en la adultez. Puede coexistir con la PASD, probablemente por mutaciones postcigóticas que causan una pérdida de la heterocigosidad (mosaicismo tipo 2)49,51.
- e.
Poroqueratosis diseminada eruptiva
 y muslo izquierdo (B). C-D. Poroqueratosis diseminada eruptiva. Múltiples placas de borde anular eritematoso e hiperqueratósico en ambos brazos (C) y en la pierna izquierda (D), las lesiones eran intensamente pruriginosas.")
A-B. Poroqueratosis lineal. Placas eritematomarronáceas de borde descamativo y trayecto lineal en el costado derecho (A) y muslo izquierdo (B). C-D. Poroqueratosis diseminada eruptiva. Múltiples placas de borde anular eritematoso e hiperqueratósico en ambos brazos (C) y en la pierna izquierda (D), las lesiones eran intensamente pruriginosas.
La PDE incluye a la poroqueratosis papular pruriginosa eruptiva y a la forma inflamatoria de poroqueratosis superficial diseminada. Es una variante muy infrecuente de poroqueratosis caracterizada por una erupción abrupta de lesiones cutáneas eritematosas e intensamente pruriginosas (fig. 2C-D). Pese a no estar claramente definida, algunos autores la subdividen en formas paraneoplásicas, asociadas con una inmunosupresión, inflamatorias u otras. Las asociadas con neoplasias pueden remitir tras el tratamiento de éstas y las variantes inflamatorias, tener resolución espontánea52,53. Hemos encontrado en la literatura 30 casos de PDE, la mayoría en hombres de mediana edad (13-84 años). Más del 50% de los pacientes había presentado previamente otras formas de poroqueratosis, con mayor frecuencia una PSD. En la histología, lo más característico es la presencia de una lamela cornoide y de un infiltrado inflamatorio dérmico (en nueve casos de predominio linfohistiocitario y en ocho, eosinofílico). Algunos autores han descrito un infiltrado inflamatorio compuesto por linfocitos T CD8+ (en la PSD lo habitual son los CD4+) y sugieren que la PDE sería una respuesta inmunológica mediada por linfocitos CD8+ contra los clones de queratinocitos anormales54,55. Lo más relevante desde el punto de vista clínico es la asociación en casi un tercio de los individuos con diversas neoplasias, principalmente hepatobiliares y hematológicas18,21,31,56-60, por lo que debería considerarse el cribado de éstas (tabla 2). Casi el 20% de los casos de PDE pueden presentar una infección vírica asociada (virus herpes simple recurrente, virus hepatitis C57 y virus hepatitis B), y existen casos relacionados con inmunosupresión61 y fármacos21. En cuanto al tratamiento, en la gran mayoría de los pacientes (75%) el cuadro se resuelve de forma espontánea en alrededor de seis meses (aunque en algunos puede tardar hasta dos años), y la respuesta al tratamiento tópico o sistémico es muy variable (nula o escasa en la mayoría de los casos).
- f.
Poroqueratosis palmo-plantar y diseminada
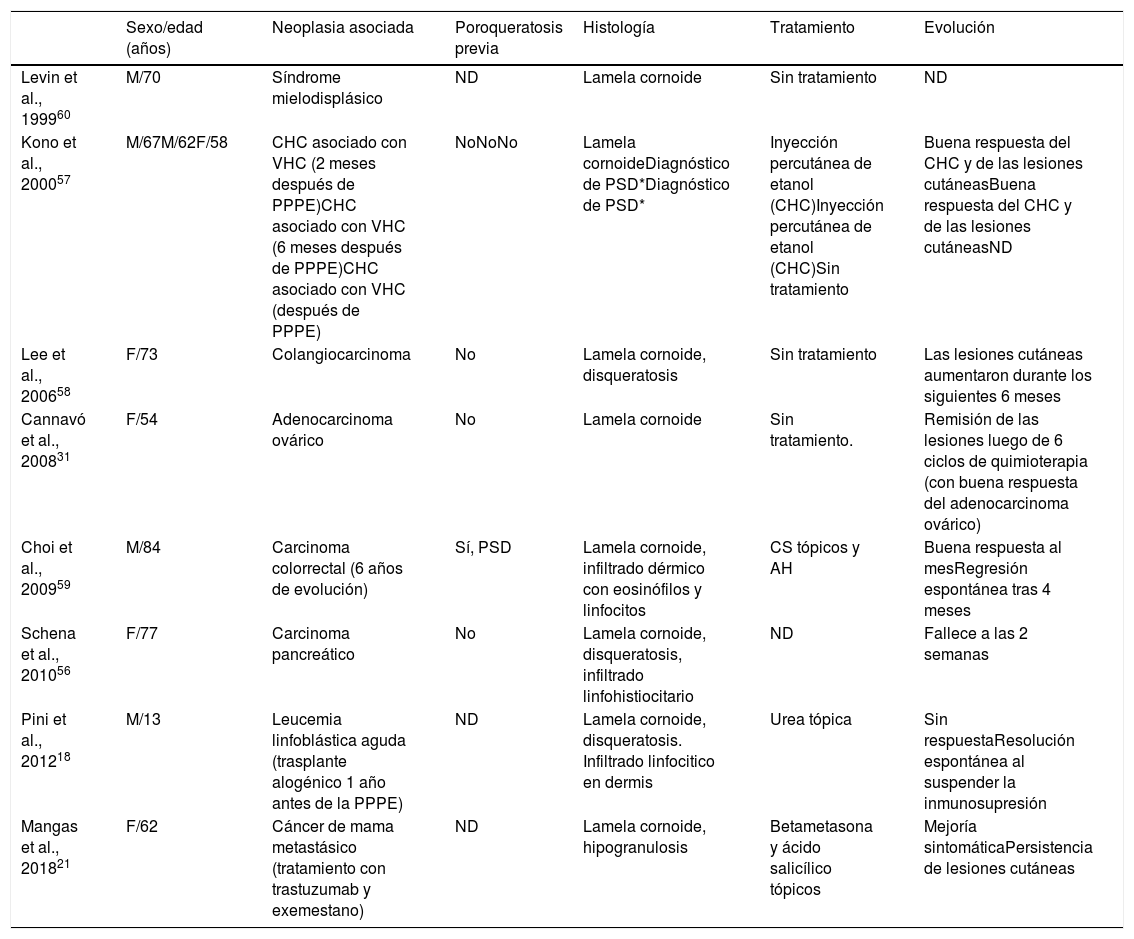
Casos descritos en la literatura de poroqueratosis diseminada eruptiva asociada con neoplasias
| Sexo/edad (años) | Neoplasia asociada | Poroqueratosis previa | Histología | Tratamiento | Evolución | |
|---|---|---|---|---|---|---|
| Levin et al., 199960 | M/70 | Síndrome mielodisplásico | ND | Lamela cornoide | Sin tratamiento | ND |
| Kono et al., 200057 | M/67M/62F/58 | CHC asociado con VHC (2 meses después de PPPE)CHC asociado con VHC (6 meses después de PPPE)CHC asociado con VHC (después de PPPE) | NoNoNo | Lamela cornoideDiagnóstico de PSD*Diagnóstico de PSD* | Inyección percutánea de etanol (CHC)Inyección percutánea de etanol (CHC)Sin tratamiento | Buena respuesta del CHC y de las lesiones cutáneasBuena respuesta del CHC y de las lesiones cutáneasND |
| Lee et al., 200658 | F/73 | Colangiocarcinoma | No | Lamela cornoide, disqueratosis | Sin tratamiento | Las lesiones cutáneas aumentaron durante los siguientes 6 meses |
| Cannavó et al., 200831 | F/54 | Adenocarcinoma ovárico | No | Lamela cornoide | Sin tratamiento. | Remisión de las lesiones luego de 6 ciclos de quimioterapia (con buena respuesta del adenocarcinoma ovárico) |
| Choi et al., 200959 | M/84 | Carcinoma colorrectal (6 años de evolución) | Sí, PSD | Lamela cornoide, infiltrado dérmico con eosinófilos y linfocitos | CS tópicos y AH | Buena respuesta al mesRegresión espontánea tras 4 meses |
| Schena et al., 201056 | F/77 | Carcinoma pancreático | No | Lamela cornoide, disqueratosis, infiltrado linfohistiocitario | ND | Fallece a las 2 semanas |
| Pini et al., 201218 | M/13 | Leucemia linfoblástica aguda (trasplante alogénico 1 año antes de la PPPE) | ND | Lamela cornoide, disqueratosis. Infiltrado linfocitico en dermis | Urea tópica | Sin respuestaResolución espontánea al suspender la inmunosupresión |
| Mangas et al., 201821 | F/62 | Cáncer de mama metastásico (tratamiento con trastuzumab y exemestano) | ND | Lamela cornoide, hipogranulosis | Betametasona y ácido salicílico tópicos | Mejoría sintomáticaPersistencia de lesiones cutáneas |
PSD, poroqueratosis superficial diseminada; PPPE, poroqueratosis papular pruriginosa eruptiva; CS, glucocorticoides; AH, antihistamínicos; VHC, virus hepatitis C; CHC, carcinoma hepatocelular; ND, no descrito.
La PPPD es una variante rara de la que existen menos de 20 casos descritos, en su mayoría familiares con un patrón de herencia autosómico dominante. Suele comenzar durante la adolescencia y afecta predominantemente a los hombres (2:1)62. Clínicamente, cursa con unas lesiones similares a PSD, con unas pápulas queratósicas de 1-2 mm, bilaterales y simétricas. En la mayoría de los casos se inicia en las áreas palmo-plantares y, tras meses o años, se extienden al resto del cuerpo62,63. Hay casos anecdóticos que se inician en el tronco y las extremidades con un compromiso palmo-plantar posterior64. Puede exacerbarse durante el periodo estival.
- g.
Poroqueratosispunctata
Para algunos autores es un subtipo de PPPD65. Debuta en la adolescencia y adultez joven, con múltiples pápulas queratósicas de 1-2 mm en las palmas y las plantas, similares a una semilla, deprimidas (hoyuelos o pits) o sobreelevadas como espículas. Suele asociarse con PM o PL65,66.
- h.
Poroqueratosis genitoglútea
Cualquier forma de poroqueratosis generalizada puede afectar a los genitales y la región glútea, sin embargo, son pocos los casos en los que se presenta exclusivamente en esta región67. Se describen tres variantes clínicas:
- 1.
Poroqueratosis clásica de la región genital. Afecta a adultos de mediana edad, con un predominio en hombres. Se ha sugerido una mayor incidencia en la población asiática y afroamericana68,69. La fricción y el rascado podrían ser un factor determinante. Se manifiesta con unas placas anulares atróficas en el centro, muy pruriginosas, distribuidas en el escroto, el pene, los glúteos y la porción proximal de los muslos68.
- 2.
Poroqueratosis ptychotropica. Denominada a partir del griego ptyhce (pliegue) y trope (torneado)70. Es una variante muy poco frecuente de poroqueratosis, con menos de 30 casos descritos en la literatura. Afecta a adultos de edad media (90% hombres)71. Se caracteriza por unas placas verrucosas gruesas eritemato-marronáceas y pruriginosas, de crecimiento lentamente progresivo, localizadas en la zona perianal, los glúteos y el pliegue interglúteo con patrón «en alas de mariposa» y la presencia de lesiones satélite (fig. 3B). También puede afectar a los genitales68. Es frecuente el retraso diagnóstico por confundirse con eccemas, condilomas acuminados o micosis71. En la microscopía se diferencia por presentar múltiples lamelas cornoides distribuidas en toda la lesión (no sólo en el borde). Puede existir un depósito de amiloide en la dermis papilar72. No se ha demostrado la presencia de agentes infecciosos en las lesiones y no parece haber una relación con una infección por el virus del papiloma humano73.
- 3.
Poroqueratosis pene-escrotal. Se ha descrito durante la tercera década, con placas granulares en el cuerpo del pene y escroto anterior, asociadas con un prurito intenso68,74. Al igual que en la variante ptychotropica, presenta múltiples lamelas cornoides74.
- i.
Poroqueratosis folicular
Descrita hace sólo una década75, hay 11 casos descritos de esta variante. Puede afectar a todas las edades, con un ligero predominio en hombres76. Se presenta como unas maculo-pápulas o placas anulares con unos bordes queratósicos, en la mayoría de los casos afectando al rostro, pero puede comprometer también el tronco y las extremidades77. En la microscopía se caracteriza por presentar la lamela cornoide sobre unos folículos dilatados75,76.
- j.
Otras variantes
Se han publicado casos aislados de poroqueratosis pustular78, ulcerativa79, ampollosa80, reticulada81, tipo prurigo nodular82 y tipo queratosis seborreica83.
Recientemente se ha descrito el poroqueratoma, que corresponde a un acantoma con múltiples lamelas cornoides. Se manifiesta como una o escasas lesiones verrucosas en las extremidades, el tronco o los glúteos84. Para algunos autores representa una variante de poroqueratosis hiperqueratósica y para otros una entidad distinta85.
Diagnóstico- a.
Clínico
A menudo el diagnóstico de poroqueratosis se basa sólo en el examen clínico, reservándose la biopsia cutánea para los casos atípicos o dudosos.
- b.
Dermatoscopia y otras pruebas de imagen
En las distintas formas clínicas se observa un borde periférico hiperqueratósico blanco, amarillo o parduzco, «en doble riel»; un área central homogénea que puede tener un aspecto cicatricial, presentar puntos o glóbulos marrones y algunas estructuras vasculares como vasos globulares y lineales irregulares que cruzan la lesión86,87 (fig. 4A-B). El uso de marcadores de tinta puede ser útil, ya que, al limpiarla con alcohol 70%, se mantiene en el borde de la lamela cornoide, diferenciándola de otras dermatosis88.
. Lesión con zonas blancas sin estructura y vasos puntiformes, con borde anular hiperqueratósico. Se puede observar el signo del «doble riel» (B) (3 Gen Dermlite DL200, x8). C-D. Histología. C. Invaginación epidérmica y presencia de columna de paraqueratosis (lamela cornoide) (flecha) (H-E, x40). D. Lamela cornoide. Nótese la ausencia de la capa granulosa en la base de la columna paraqueratósica (flecha), junto a la presencia de queratinocitos disqueratósicos en el estrato espinoso (H-E, x200). Lamela cornoide (flecha) (H-E, x100).")
Poroqueratosis. A-B. Dermatoscopia. Lesión eritematomarronácea sin estructuras claramente definidas con borde anular marronáceo (A). Lesión con zonas blancas sin estructura y vasos puntiformes, con borde anular hiperqueratósico. Se puede observar el signo del «doble riel» (B) (3 Gen Dermlite DL200, x8). C-D. Histología. C. Invaginación epidérmica y presencia de columna de paraqueratosis (lamela cornoide) (flecha) (H-E, x40). D. Lamela cornoide. Nótese la ausencia de la capa granulosa en la base de la columna paraqueratósica (flecha), junto a la presencia de queratinocitos disqueratósicos en el estrato espinoso (H-E, x200). Lamela cornoide (flecha) (H-E, x100).
Al utilizar la microscopía confocal de reflectancia la lamela cornoide se observa como una estructura paraqueratótica brillante, sin capa granular subyacente87. También es posible observar la lamela cornoide con tomografía de coherencia óptica2.
- c.
Histología
Al hacer una biopsia el borde de una lesión de poroqueratosis, se puede observar una columna de células paraqueratósicas bien delimitada (lamela cornoide). En esta zona hay hipo/agranulosis y, ocasionalmente, unas células disqueratóticas o queratinocitos vacuolados (fig. 4C-D). Se ha descrito la presencia de una espongiosis eosinofílica en algunos casos aislados. En la dermis se puede observar un escaso infiltrado inflamatorio perivascular, compuesto principalmente por linfocitos CD4+1,2. En la PDE se ha observado un infiltrado de linfocitos CD8+55. En la zona central de las lesiones existe una hiperqueratosis ortoqueratósica leve1.
Diagnóstico diferencialLa tabla 3 resume los múltiples diagnósticos diferenciales que hay considerar en las distintas variables de la poroqueratosis1,2,46,66,89. La principal característica histopatológica de la poroqueratosis es la presencia de la lamela cornoide. Sin embargo, existen varias patologías en las que ésta se puede observar, tales como las verrugas vulgares, las queratosis actínicas, algunas ictiosis y la hiperqueratosis nevoide. En la poroqueratosis suele estar ausente la capa granulosa bajo la paraqueratosis2. También es posible observar la lamela cornoide en la queratosis punctata hereditaria y la queratodermia espinulosa, siendo difícil diferenciarlas de la PPPD y la PP. Una clave diagnóstica es que en estas variantes de poroqueratosis suelen observarse disqueratocitos o vacuolización de algunos queratinocitos66. Además, se observan múltiples lamelas cornoides en el poroqueratoma y la poroqueratosis genitoglútea71,84.
Diagnóstico diferencial de la poroqueratosis
| Subtipo de poroqueratosis | Diagnóstico diferencial |
|---|---|
| PSD y PASD | Queratosis actínicas, queratosis seborreicas, liquen plano, tiña corporis, psoriasis, eccema numular y pitiriasis rosada. |
| PM | Enfermedad de Bowen, granuloma anular, hipoqueratosis palmar/plantar circunscrita, liquen simple crónico, tiña corporis, sarcoidosis |
| PL | Verrugas lineales, liquen estriado, nevus epidérmico verrucoso lineal, enfermedad de Darier lineal y nevus poroqueratósico del ostium y conducto dérmico ecrinos. |
| PPPD y PP | Queratosis punctata hereditaria, queratodermia espinulosa, síndrome de Gorlin, enfermedad de Darier, queratosis arsenical y liquen nitidus palmo-plantar. |
| Poroqueratosis ptychotropica | Psoriasis, acrodermatitis enteropática y eritema necrolítico migratorio |
PSD, poroqueratosis actínica superficial diseminada; PASD, poroqueratosis actínica superficial diseminada; PM, poroqueratosis de Mibelli; PL, poroqueratosis lineal; PPPD, poroqueratosis palmo-plantar y diseminada; PP, poroqueratosis punctata.
Fuente: Kanitakis et al.1, Sertznig et al.2, Ferreira et al.46, Teixeira et al.66, Elfatoiki et al.89.
La poroqueratosis tiende a tener un curso crónico y a ser refractaria al tratamiento, reemplazar por “refractaria al tratamiento, y sólo un 16% de los casos muestran una respuesta completa4. Por esto, una opción válida en pacientes asintomáticos o con lesiones limitadas es la conducta expectante, que incluye la recomendación de realizar una fotoprotección adecuada, el uso de emolientes y la realización de controles regulares durante un tiempo prolongado (fig. 5). Esto último es muy importante dado el riesgo de malignización (especialmente en PM, PL y PPPD), en especial en las lesiones de gran tamaño1. En el caso de la PDE, es recomendable la búsqueda exhaustiva de una neoplasia subyacente52, principalmente de origen hepatobiliopancreático o hematológico.
.")
Algoritmo de tratamiento de las poroqueratosis.
PDE, poroqueratosis diseminada eruptiva; PSD, poroqueratosis superficial diseminada; PASD, poroqueratosis actínica superficial diseminada; PM, poroqueratosis de Mibelli; PPPD, poroqueratosis palmo-plantar y diseminada; PL, poroqueratosis lineal; TFD terapia fotodinámica; vit, vitamina; 5-FU, 5-fluorouracilo al 5%; RTG, radioterapia con rayos Grenz.
*Estudio histopatológico sólo en casos atípicos o dudosos.
**Principalmente biliohepatopancreáticas y hematológicas. Solicitar hemograma, pruebas hepáticas, LDH, tomografía tóraco-abdominal.
***Descartar malignización cutánea (carcinoma escamoso y carcinoma basocelular).
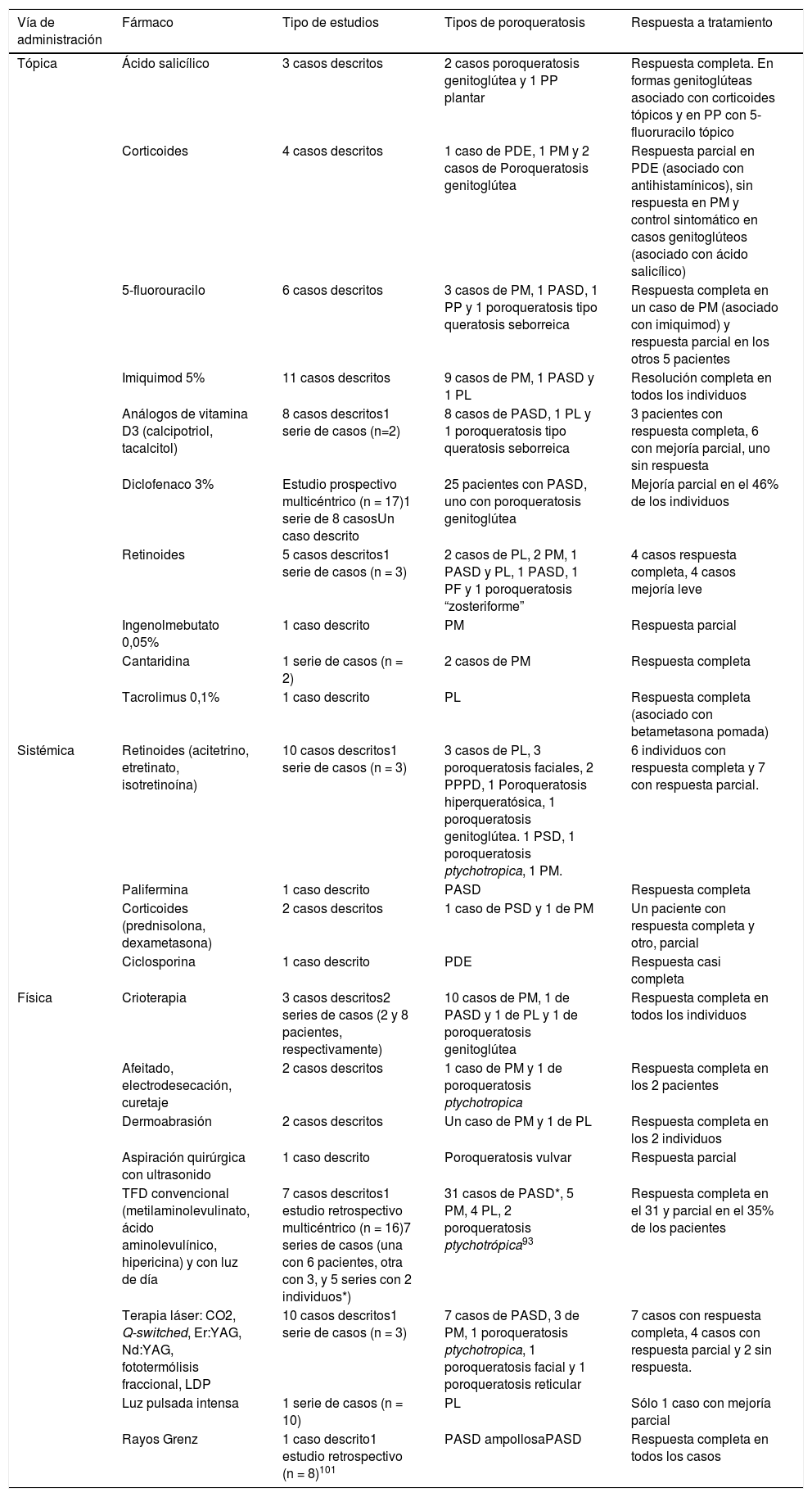
En el caso de las lesiones sintomáticas, no existen ensayos clínicos controlados que avalen la eficacia de las distintas opciones terapéuticas. Las recomendaciones están basadas principalmente en descripciones de casos, series pequeñas con menos de 20 pacientes y dos revisiones sistemáticas de la literatura90,91. La tabla 4 resume las opciones terapéuticas tópicas, sistémicas y físicas, y su grado de evidencia. En las lesiones de menor tamaño y sintomáticas, se puede realizar crioterapia92, curetaje, electrocirugía, terapia fotodinámica o exéresis quirúrgica, con el riesgo de hiperpigmentación postinflamatoria o formación de cicatrices90,93. En las lesiones de mayor tamaño o múltiples, pero localizadas, se pueden indicar análogos tópicos de vitamina D, retinoides tópicos, 5-fluoruracilo o imiquimod 5%90,94. En la PM pareciera que el imiquimod puede ser la mejor opción90. En la PL se podrían utilizar retinoides tópicos u orales como primera opción terapéutica90. La TFD podría recomendarse en primera o segunda línea en las formas localizadas de poroqueratosis.
Tratamiento de la poroqueratosis y su respuesta clínica
| Vía de administración | Fármaco | Tipo de estudios | Tipos de poroqueratosis | Respuesta a tratamiento |
|---|---|---|---|---|
| Tópica | Ácido salicílico | 3 casos descritos | 2 casos poroqueratosis genitoglútea y 1 PP plantar | Respuesta completa. En formas genitoglúteas asociado con corticoides tópicos y en PP con 5-fluoruracilo tópico |
| Corticoides | 4 casos descritos | 1 caso de PDE, 1 PM y 2 casos de Poroqueratosis genitoglútea | Respuesta parcial en PDE (asociado con antihistamínicos), sin respuesta en PM y control sintomático en casos genitoglúteos (asociado con ácido salicílico) | |
| 5-fluorouracilo | 6 casos descritos | 3 casos de PM, 1 PASD, 1 PP y 1 poroqueratosis tipo queratosis seborreica | Respuesta completa en un caso de PM (asociado con imiquimod) y respuesta parcial en los otros 5 pacientes | |
| Imiquimod 5% | 11 casos descritos | 9 casos de PM, 1 PASD y 1 PL | Resolución completa en todos los individuos | |
| Análogos de vitamina D3 (calcipotriol, tacalcitol) | 8 casos descritos1 serie de casos (n=2) | 8 casos de PASD, 1 PL y 1 poroqueratosis tipo queratosis seborreica | 3 pacientes con respuesta completa, 6 con mejoría parcial, uno sin respuesta | |
| Diclofenaco 3% | Estudio prospectivo multicéntrico (n = 17)1 serie de 8 casosUn caso descrito | 25 pacientes con PASD, uno con poroqueratosis genitoglútea | Mejoría parcial en el 46% de los individuos | |
| Retinoides | 5 casos descritos1 serie de casos (n = 3) | 2 casos de PL, 2 PM, 1 PASD y PL, 1 PASD, 1 PF y 1 poroqueratosis “zosteriforme” | 4 casos respuesta completa, 4 casos mejoría leve | |
| Ingenolmebutato 0,05% | 1 caso descrito | PM | Respuesta parcial | |
| Cantaridina | 1 serie de casos (n = 2) | 2 casos de PM | Respuesta completa | |
| Tacrolimus 0,1% | 1 caso descrito | PL | Respuesta completa (asociado con betametasona pomada) | |
| Sistémica | Retinoides (acitetrino, etretinato, isotretinoína) | 10 casos descritos1 serie de casos (n = 3) | 3 casos de PL, 3 poroqueratosis faciales, 2 PPPD, 1 Poroqueratosis hiperqueratósica, 1 poroqueratosis genitoglútea. 1 PSD, 1 poroqueratosis ptychotropica, 1 PM. | 6 individuos con respuesta completa y 7 con respuesta parcial. |
| Palifermina | 1 caso descrito | PASD | Respuesta completa | |
| Corticoides (prednisolona, dexametasona) | 2 casos descritos | 1 caso de PSD y 1 de PM | Un paciente con respuesta completa y otro, parcial | |
| Ciclosporina | 1 caso descrito | PDE | Respuesta casi completa | |
| Física | Crioterapia | 3 casos descritos2 series de casos (2 y 8 pacientes, respectivamente) | 10 casos de PM, 1 de PASD y 1 de PL y 1 de poroqueratosis genitoglútea | Respuesta completa en todos los individuos |
| Afeitado, electrodesecación, curetaje | 2 casos descritos | 1 caso de PM y 1 de poroqueratosis ptychotropica | Respuesta completa en los 2 pacientes | |
| Dermoabrasión | 2 casos descritos | Un caso de PM y 1 de PL | Respuesta completa en los 2 individuos | |
| Aspiración quirúrgica con ultrasonido | 1 caso descrito | Poroqueratosis vulvar | Respuesta parcial | |
| TFD convencional (metilaminolevulinato, ácido aminolevulínico, hipericina) y con luz de día | 7 casos descritos1 estudio retrospectivo multicéntrico (n = 16)7 series de casos (una con 6 pacientes, otra con 3, y 5 series con 2 individuos*) | 31 casos de PASD*, 5 PM, 4 PL, 2 poroqueratosis ptychotrópica93 | Respuesta completa en el 31 y parcial en el 35% de los pacientes | |
| Terapia láser: CO2, Q-switched, Er:YAG, Nd:YAG, fototermólisis fraccional, LDP | 10 casos descritos1 serie de casos (n = 3) | 7 casos de PASD, 3 de PM, 1 poroqueratosis ptychotropica, 1 poroqueratosis facial y 1 poroqueratosis reticular | 7 casos con respuesta completa, 4 casos con respuesta parcial y 2 sin respuesta. | |
| Luz pulsada intensa | 1 serie de casos (n = 10) | PL | Sólo 1 caso con mejoría parcial | |
| Rayos Grenz | 1 caso descrito1 estudio retrospectivo (n = 8)101 | PASD ampollosaPASD | Respuesta completa en todos los casos |
PM, poroqueratosis de Mibelli; PASD, poroqueratosis actínica superficial diseminada; PP, poroqueratosis punctata; PDE, poroqueratosis diseminada eruptiva; PF, poroqueratosis folicular; TFD, terapia fotodinámica; LPD, láser decolorante pulsado; Er:YAG, láser de erbio Yag;Nd:Yag, láser de neodimio Yag
En el caso de una enfermedad diseminada como la PASD o PSD, existen múltiples descripciones de casos aislados con unas respuestas buenas con los análogos de la vitamina D (AVD), con muy escasos o nulos efectos adversos95. Por esta razón, algunos autores lo recomiendan como una opción de primera línea en estos casos90. En un estudio prospectivo de 17 adultos con PASD, el mayor realizado en pacientes con poroqueratosis, se utilizó el diclofenaco tópico al 3% durante 3-6 meses y se evitó la progresión de las lesiones en más del 50% de los casos96. Sin embargo, en otra serie de ocho pacientes tratados con diclofenaco 3%, sólo se observó una respuesta parcial en dosindividuos97. En cuanto a las terapias físicas, la terapia fotodinámica, tanto convencional como con luz de día, es una de las opciones para la que existe un mayor número de casos descritos (alrededor de 42), con unas tasas de respuesta completa o parcial del 31 y 35% de los individuos, respectivamente (sobre todo en PASD)90,91,98,99, pero puede ocasionar considerables efectos adversos91. Diversas formas de terapia láser han sido utilizadas en un número muy limitado de pacientes (alrededor de 13 individuos) con resultados prometedores, con buenas tasas de respuesta y escasos efectos adversos91. La luz pulsada intensa, pese a ser útil en múltiples dermatosis, no demostró ser beneficiosa en una serie de 10 pacientes (uno de ellos con respuesta completa) con PSD100. Otra forma de terapia física utilizada ha sido la radioterapia con rayos Grenz (RTG). En un estudio retrospectivo de ocho pacientes con RTG (6 a 10 sesiones, dosis total de 28 a 52 Gy), todos alcanzaron una respuesta completa. Sólo un individuo debió suspender la RTG por efectos adversos, y sólo un paciente recidivó. Ninguno presentó un carcinoma cutáneo en la superficie irradiada durante el seguimiento clínico101. En cuanto a la terapia sistémica, se han utilizado retinoides orales en 13 pacientes con una respuesta completa en seis de ellos y respuesta parcial en siete102. Se han usado los glucocorticoides sistémicos en dos individuos, uno de ellos con respuesta completa)2,90, y la ciclosporina en un caso con una respuesta casi completa103.
ConclusiónLas poroqueratosis son dermatosis infrecuentes, con un amplio espectro de variantes clínicas que se deben conocer para evitar errores y retraso diagnóstico. Cobra relevancia el seguimiento a largo plazo para la detección precoz de malignización cutánea, un riesgo presente en todas las variantes de poroqueratosis. En las formas eruptivas se debe descartar la asociación con neoplasias viscerales o hematológicas. El tratamiento de las poroqueratosis es complejo y, a menudo, insatisfactorio, y la conducta expectante puede ser una opción para tener en cuenta.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses



