






Mycosis fungoides (MF), the most common primary cutaneous T-cell lymphoma, is unusual in children.
ObjectivesWe aimed to describe the epidemiologic, clinical, histopathologic, and immunophenotypic characteristics of MF as well as treatments and course of disease in a pediatric case series.
Material and methodsData for all patients admitted to our pediatric hospital (Hospital Dr. J. P. Garrahan) in Argentina with a clinical and histopathologic diagnosis of MF between August 1988 and July 2014 were included.
ResultsA total of 14 patients were diagnosed with MF. The ratio of boys to girls was 1:1.33. The mean age at diagnosis was 11.23 years (range, 8–15 years). The mean time between onset and diagnosis was 3.5 years (range, 4 months–7 years). All patients had hypopigmented MF and 42% also presented the features of classic MF. Seven (50%) had the CD8+ immunophenotype exclusively. Seventy-eight percent were in stage IB at presentation. Phototherapy was the treatment of choice. Four patients relapsed at least once and skin lesions progressed in 3 patients. All patients improved.
ConclusionsMF is unusual in children. The hypopigmented form is the most common. Diagnosis is delayed because the condition is similar to other hypopigmented diseases seen more often in childhood. Although prognosis is good, the rate of recurrence is high, so long-term follow-up is necessary.
La micosis fungoide (MF) es el linfoma cutáneo primario de células T más frecuente. Su aparición en la infancia es excepcional.
ObjetivosDescribir las características epidemiológicas, clínicas, histopatológicas e inmunofenotípicas de los pacientes con MF. Describir los tratamientos utilizados y la evolución.
Material y métodoSe incluyeron todos los pacientes admitidos en el Hospital de Pediatría Dr. J. P. Garrahan (Argentina) en el período comprendido entre agosto de 1988 y julio de 2014 con diagnóstico clínico e histopatológico de MF.
ResultadosSe diagnosticaron 14 pacientes con MF. La distribución por sexo fue M/F: 1:1,33. La edad media al diagnóstico fue de 11,23 años (rango: 8 a 15 años). El tiempo promedio de evolución hasta el momento del diagnóstico fue de 3 años y 6 meses (rango: 4 meses a 7 años). Todos los pacientes presentaron la forma clínica hipopigmentada y en el 42% se asoció la forma clásica. El 50% (n=7) exhibió un inmunofenotipo CD8 positivo de forma exclusiva. El 78% presentó estadio IB. La fototerapia fue el tratamiento de elección. Cuatro pacientes tuvieron por lo menos una recaída y 3 demostraron progresión de su enfermedad a nivel cutáneo. La evolución fue favorable en todos los casos.
ConclusionesLa MF es una entidad infrecuente en la infancia, siendo la forma hipopigmentada la más frecuente. Su diagnóstico es tardío debido a la similitud con otras enfermedades hipopigmentadas frecuentes en la niñez. A pesar de tener un buen pronóstico, presenta alta tasa de recidivas y requiere un seguimiento a largo plazo.
Mycosis fungoides (MF) is the most common primary T-cell cutaneous lymphoma and it mainly affects adults in the fifth decade of life.1 Clinically, it has 3 classic stages (macule, plaque, and tumor) that can overlap. Patients typically have exclusive cutaneous involvement that follows an indolent course for years or even decades. Characteristic histopathologic findings include an epidermotropic tumor infiltrate and proliferation of atypical T cells with a cerebriform appearance.
Primary cutaneous lymphomas are rare in childhood. MF is also rare, but, as with adults, it is the most common type of primary cutaneous lymphoma in this population.2 The first descriptions of MF in children date back to 1984.3 Several cases have been reported since and the most common clinical presentation in most cases has been hypopigmented MF.2,4–11 Histopathologic findings are similar to those seen in adults, but immunohistochemical studies show a predominant CD8+ T-cell infiltrate, which would correlate with the hypopigmented clinical lesions.
The aim of this study was to describe the epidemiological, clinical, histopathologic, and immunophenotypic characteristics of MF in pediatric patients and report on T-cell receptor (TCR) gene rearrangement test results, treatments, and disease course.
Patients and MethodsWe conducted a retrospective, observational, descriptive, cross-sectional study of patients with a clinical and histopathologic diagnosis of MF seen at Hospital de Pediatría S.A.M.I.C. «Prof. Dr. Juan P. Garrahan» in Buenos Aires, Argentina between August 1988 and July 2004. This hospital is a referral hospital for children in our country and Latin America. The following variables were analyzed: sex, age, initial diagnosis, time from onset to diagnosis, clinical manifestations, location, stage at diagnosis, histopathologic findings, immunophenotype, TCR gene rearrangement, treatments received, disease course, and follow-up duration. We reviewed clinical charts and pathology reports and applied the diagnostic criteria of the World Health Organization/European Organisation for Research and Treatment of Cancer (2005)1 and the staging criteria of the International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer (2007).12
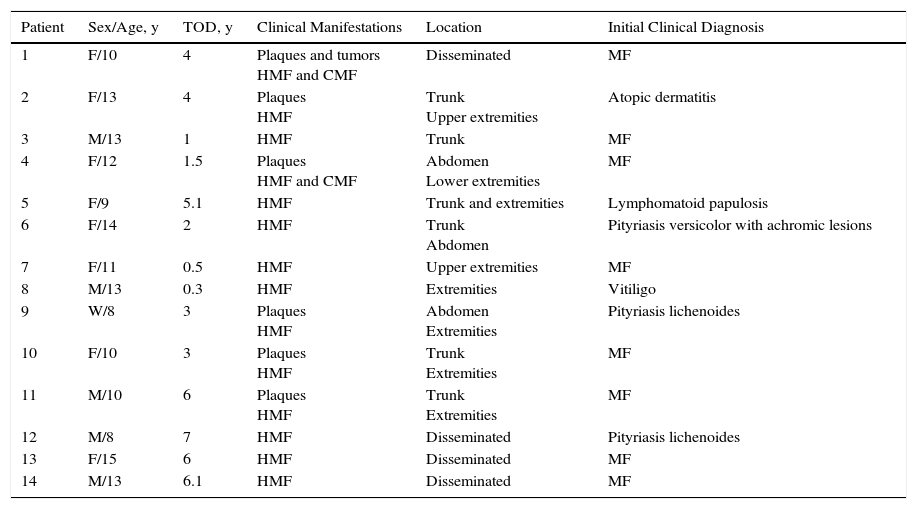
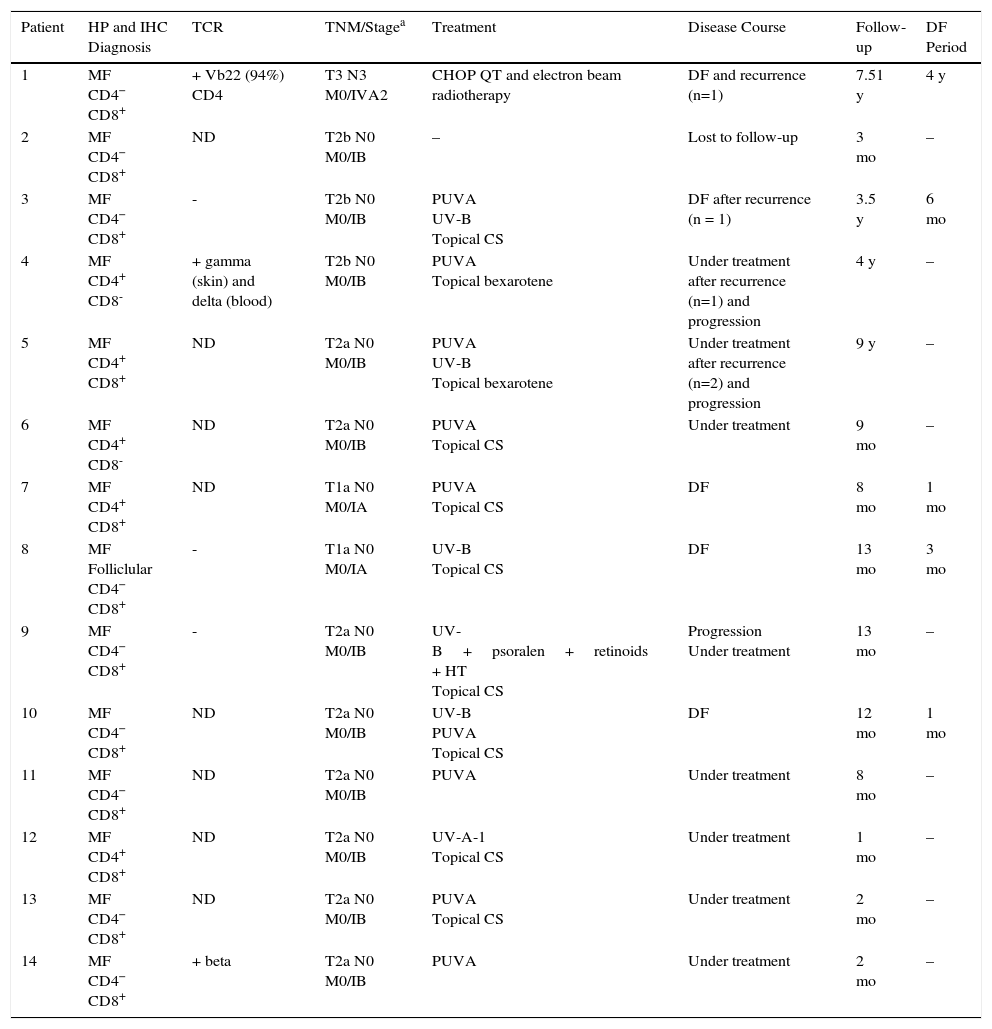
ResultsFourteen patients with clinical and histologic features of MF were included in the study. The male to female ratio was 1:1.33. Mean age at diagnosis was 11.23 years (range, 8-15 years) and mean time from onset to diagnosis was 3 years and 6 months (range, 4 months to 7 years). Information on demographic, clinical, histopathologic, and immunohistochemical characteristics, TCR gene rearrangement, disease stage, treatments, disease course, follow-up, and disease-free months is summarized in Tables 1 and 2.
Clinical Data.
| Patient | Sex/Age, y | TOD, y | Clinical Manifestations | Location | Initial Clinical Diagnosis |
|---|---|---|---|---|---|
| 1 | F/10 | 4 | Plaques and tumors HMF and CMF | Disseminated | MF |
| 2 | F/13 | 4 | Plaques HMF | Trunk Upper extremities | Atopic dermatitis |
| 3 | M/13 | 1 | HMF | Trunk | MF |
| 4 | F/12 | 1.5 | Plaques HMF and CMF | Abdomen Lower extremities | MF |
| 5 | F/9 | 5.1 | HMF | Trunk and extremities | Lymphomatoid papulosis |
| 6 | F/14 | 2 | HMF | Trunk Abdomen | Pityriasis versicolor with achromic lesions |
| 7 | F/11 | 0.5 | HMF | Upper extremities | MF |
| 8 | M/13 | 0.3 | HMF | Extremities | Vitiligo |
| 9 | W/8 | 3 | Plaques HMF | Abdomen Extremities | Pityriasis lichenoides |
| 10 | F/10 | 3 | Plaques HMF | Trunk Extremities | MF |
| 11 | M/10 | 6 | Plaques HMF | Trunk Extremities | MF |
| 12 | M/8 | 7 | HMF | Disseminated | Pityriasis lichenoides |
| 13 | F/15 | 6 | HMF | Disseminated | MF |
| 14 | M/13 | 6.1 | HMF | Disseminated | MF |
Abbreviations: CMF, classic mycosis fungoides; F, female; HMF, hypopigmented mycosis fungoides; M, male; TOD, time from onset to diagnosis.
Clinical Findings, Treatment, and Disease Course.
| Patient | HP and IHC Diagnosis | TCR | TNM/Stagea | Treatment | Disease Course | Follow-up | DF Period |
|---|---|---|---|---|---|---|---|
| 1 | MF CD4− CD8+ | + Vb22 (94%) CD4 | T3 N3 M0/IVA2 | CHOP QT and electron beam radiotherapy | DF and recurrence (n=1) | 7.51 y | 4 y |
| 2 | MF CD4− CD8+ | ND | T2b N0 M0/IB | – | Lost to follow-up | 3 mo | – |
| 3 | MF CD4− CD8+ | - | T2b N0 M0/IB | PUVA UV-B Topical CS | DF after recurrence (n = 1) | 3.5 y | 6 mo |
| 4 | MF CD4+ CD8- | + gamma (skin) and delta (blood) | T2b N0 M0/IB | PUVA Topical bexarotene | Under treatment after recurrence (n=1) and progression | 4 y | – |
| 5 | MF CD4+ CD8+ | ND | T2a N0 M0/IB | PUVA UV-B Topical bexarotene | Under treatment after recurrence (n=2) and progression | 9 y | – |
| 6 | MF CD4+ CD8- | ND | T2a N0 M0/IB | PUVA Topical CS | Under treatment | 9 mo | – |
| 7 | MF CD4+ CD8+ | ND | T1a N0 M0/IA | PUVA Topical CS | DF | 8 mo | 1 mo |
| 8 | MF Folliclular CD4− CD8+ | - | T1a N0 M0/IA | UV-B Topical CS | DF | 13 mo | 3 mo |
| 9 | MF CD4− CD8+ | - | T2a N0 M0/IB | UV-B+psoralen+retinoids + HT Topical CS | Progression Under treatment | 13 mo | – |
| 10 | MF CD4− CD8+ | ND | T2a N0 M0/IB | UV-B PUVA Topical CS | DF | 12 mo | 1 mo |
| 11 | MF CD4− CD8+ | ND | T2a N0 M0/IB | PUVA | Under treatment | 8 mo | – |
| 12 | MF CD4+ CD8+ | ND | T2a N0 M0/IB | UV-A-1 Topical CS | Under treatment | 1 mo | – |
| 13 | MF CD4− CD8+ | ND | T2a N0 M0/IB | PUVA Topical CS | Under treatment | 2 mo | – |
| 14 | MF CD4− CD8+ | + beta | T2a N0 M0/IB | PUVA | Under treatment | 2 mo | – |
Abbreviations: CHOP,cyclophosphamide, doxorubicin, vincristine, and prednisone; CS, corticosteroids; HT, heliotherapy; DF, disease-free; IHC, immunohistochemical; MF, micosis fungoides; ND, not done; QT, chemotherapy; TCR, T-cell receptor gene rearrangement.
MF was the initial diagnosis in just 8 patients (57%). The initial diagnoses in the other 6 patients were pityriasis lichenoides chronica (2 patients), lymphomatoid papulosis (1 patient), hypopigmented pityriasis versicolor (1 patient), vitiligo (1 patient), and atopic dermatitis (1 patient) (Table 1).
All patients had hypopigmented macules (Figs. 1 and 2). Six patients (42.8%) developed concomitant or subsequent classic MF lesions. These took the form of macules (Fig. 3) in 2 patients, plaques (Fig. 4) in 6, and a tumor (Fig. 5) in 1. One patient (#9) had clinical manifestations of pityriasis lichenoides chronica but a histopathologic diagnosis of MF.





Two patients (#11 and #12) with a clinical and histopathologic diagnosis of pityriasis lichenoides chronica developed MF after a period of 7 years, and another (#5) experienced progression of lymphomatoid papulosis to MF after a period of 5 years.
All the patients had histopathologic findings consistent with MF (epidermotropic atypical lymphocytic infiltrate) and half of them had Pautrier microabscesses (Fig. 6). One of these (patient #8) had a folliculotropic histopathologic variant, but there was no correlation with clinical findings. In the immunohistochemical studies, 7 patients (50%) had a CD8+CD4– immunophenotype, 5 had a CD8+ CD4+ immunophenotype, and just 2 had a CD8– CD4+ immunophenotype (Table 2).
.")
TCR gene rearrangement tests were performed in just 6 patients. The results were positive in patient #1 (Vb22 CD4, variable region of beta chain), patient #4 (gamma), and patient #14 (beta) and negative in patients #3, #8, and #9. The studies were performed using skin samples in all cases. Blood samples were also tested in 1 patient, and the result was positive for TCR delta (Table 2).
At the time of MF diagnosis, 11 patients (78.5%) had stage IB disease, 2 had stage IA disease, and 1 had stage IVA2 disease (Table 2). The patient with stage IV disease had extracutaneous involvement of the axillary and inguinal lymph nodes.
Lactate dehydrogenase (LDH) levels and eosinophil counts were available for 11 of the 14 patients. None had elevated LDH levels and just 1 (#4) had hypereosinophilia (>700/mm3). Although this patient had chronic bronchitis episodes, it is noteworthy that he did not have hypereosinophilia during his time free of MF.
The treatments reported are summarized in Table 2, and it is important to note that some treatments were administered concurrently or sequentially in different patients. The initial treatment of choice was phototherapy in 12 patients (85.7%) due to the extension of their lesions. Six received psoralen plus UV-A (PUVA) therapy, 2 received narrowband UV-B therapy, and 3 received these treatments consecutively. Finally, 1 patient was treated with UV-A1 phototherapy.
Additional concomitant and/or consecutive treatments in 9 patients were high-potency topical corticosteroids (clobetasol) (n=6), topical bexarotene (n=2), and a combination of oral retinoids (acitretin), psoralens, and heliotherapy (n=1). The patient with stage IV disease was treated with CHOP chemotherapy (cyclophosphamide 750mg/m2/d, doxorubicin 50mg/m2/d, intravenous vincristine 1.4mg/m2/d, and methylprednisone 60mg/m2/d for 5 days) and with electron beam radiotherapy.
Eight patients are currently receiving treatment, 4 are free of disease, 1 (patient #1) was referred to an adult hospital after a disease-free period of 4 years, and another (#2) was lost to follow up.
Four of the 14 patients (#1, #3, #4, and #5) had at least 1 recurrence, 3 experienced extension of cutaneous involvement, and 1 showed partial response with persistence of MF. There were no deaths over a median follow-up period of 3 years (range, 2 months to 9 years). The median disease-free period was 3 months (range, 1-48 months). These wide ranges can be explained by the fact that 9 of 14 patients were added to the series in the last 18 months. Six of them had been followed for less than 1 year and most were on their first treatment, preventing us from evaluating overall treatment effectiveness.
DiscussionChildren account for between 5% and 16% of all MF patients, although rates vary greatly from study to study.6–9,11,13 The incidence of MF before the age of 20 years is approximately 0.05 cases per 100 000 inhabitants per year14 and MF is the most common primary cutaneous T-cell lymphoma in children. Although a predilection for the male sex has been reported,13,14 there was a slight female predominance in our series. The mean age at diagnosis (11.23 years) and the mean time from onset to diagnosis (3 years and 6 months) are similar to those reported in other publications.4,6–10,13,15
MF is difficult to diagnosis on purely clinical grounds in children due to the rarity of the disease in this population, the low index of suspicion among physicians, and the broad differential diagnosis.4,9 This diagnostic difficulty is reflected in our study by the time it took to establish a diagnosis (3 years and 6 months) and the low level of clinical suspicion at the first visit, where just 57% of children were diagnosed with MF.
Hypopigmented MF is the most common clinical variant of MF in children, and is particularly frequent in patients with Fitzpatrick skin types iii or iv.4,5,7,8,10,11,13,16,17 Forty percent of our patients had hypopigmented lesions coexisting with classic MF lesions, supporting previous reports.6,8,10 It should be highlighted that 1 patient in our series had a clinical diagnosis of pityriasis lichenoides chronica but histopathologic findings consistent with MF. Another 2 patients, one with a diagnosis of pityriasis lichenoides chronica and the other with a diagnosis of lymphomatoid papulosis developed MF after several years. These situations have been described infrequently in the literature.
Classic MF typically has a CD3+, CD4, CD5+, and CD8− immunophenotype, with loss of CD7 expression. CD8+ predominance is common in hypopigmented MF5,7,9 and was observed in 12 of our patients. It should be noted that 5 patients (35%) had a CD4+ and CD8+ immunophenotype, which is a rare variant.18,19
It is difficult to assess treatment response in our series due to the retrospective nature of our study and the fact that a majority of the children are still undergoing treatment. Sixty-four percent of patients were included in the last 18 months and almost half have been monitored for less than a year.
Treatments for MF vary according to disease stage. Because most patients have generalized cutaneous lesions, without systemic involvement, the treatment of choice is phototherapy. Topical corticosteroids are used in cases of localized disease. Phototherapy was also the treatment of choice in most of our patients. PUVA was more effective than narrowband UV-B therapy, and no adverse effects have been observed to date. Young patients with MF, however, require multiple treatments over a lifetime due to the high recurrence rates. Accordingly, prolonged maintenance phototherapy is recommended to prevent early relapse.5,13,14,17 However, while these treatment regimens prolong periods of remission, they can result in high cumulative doses of UV-A and long-term monitoring is therefore necessary due to the risk of early carcinogenesis.16 Over half of the patients in our series were treated with high-potency topical corticosteroids and/or topical bexarotene. Response was moderate or short-lasting.
Coinciding with reports in the literature,2,4,6,8–11,13,15,16 all but 1 patient in our series had stage IA or IB disease; 78% had stage IB disease, which has an excellent prognosis and a life expectancy comparable to that of the general population.1,20 Most of the patients progressed very favorably. There were no deaths and only 20% experienced disease progression. Progression, however, was marked by an extension in cutaneous involvement and none of the patients developed systemic disease. The only patient with skin tumors and lymph node involvement was free of disease after 7 years’ follow-up. Other series of childhood MF have reported similar progression-free results.5,13,15
Our study has limitations inherent to its retrospective design. We used empirical definitions to assess treatment response as there are no standardized criteria for the pediatric population as such, these definitions mainly depend on the assest of the attending specialist. Another limitation is the short follow-up period, as this made it difficult to draw conclusions on long-term outcomes, although the literature reports low rates of disease progression and excellent survival.5,9,11,13,14 Nonetheless, in a study of patients under 30 years old with MF who were followed for 10 years Ai et al.14 detected an increased risk of a second cancer (standard incidence ratio of 3.49), in particular lymphoma and melanoma. Overall survival, however, was between 88.9% and 94.3% and the differences were not statistically significant
In conclusion, MF is uncommon in children and difficult to diagnose due to the broad differential diagnosis. The hypopigmented variant is the most common form of MF in this age group. Although MF in childhood has a good prognosis, it has high recurrence rates and therefore requires long-term follow-up. Monitoring of children with MF over time will provide more insights into disease course and progression, long-term treatment effects, and onset of second cancers.
Ethical DisclosuresProtection of humans and animalsThe authors declare that no tests were carried out in humans or animals for the purpose of this study.
Confidentiality of dataThe authors declare that they have followed their hospital's protocol on the publication of data concerning patients.
Right to privacy and informed consentThe authors declare that no private patient data appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Dr Guillermo Chantada and Centro Médico PSORIAHUE.
Please cite this article as: Cervini AB, Torres-Huamani AN, Sanchez-La-Rosa C, Galluzzo L, Solernou V, Digiorge J, et al. Micosis fungoide. Experiencia en un hospital pediátrico. Actas Dermosifiliogr. 2017;108:564–570.


