



Juvenile xanthogranulomas (JXGs) are rare, benign lesions that belong to the large group of non-Langerhans cell histiocytoses. JXG presents with 1 or more erythematous or yellowish nodules that are usually located on the head or neck. Most JXG lesions are congenital or appear during the first year of life. Extracutaneous involvement is rare, but the literature traditionally suggests investigating the possibility of ocular compromise. JXG is mainly a clinical diagnosis, but a skin biopsy may sometimes be needed for confirmation. JXGs on the skin are self-limiting and usually do not require treatment. This review describes the clinical and therapeutic aspects of JXG, emphasizing available evidence and the diagnosis of extracutaneous involvement.
El xantogranuloma juvenil es un trastorno benigno poco frecuente, que pertenece al amplio grupo de las histiocitosis de células no Langerhans. Se presenta con uno o más nódulos eritematosos o amarillentos, ubicados preferentemente en la cabeza y el cuello. La mayoría de los casos se inician durante el primer año de vida, incluyendo lesiones congénitas. La afectación extracutánea es rara, sugiriéndose tradicionalmente en la literatura estudiar el compromiso ocular. El diagnóstico del xantogranuloma juvenil es fundamentalmente clínico, sin embargo, en ocasiones se requiere confirmarlo con biopsia de piel. Las lesiones cutáneas son autolimitadas, por lo que suelen no requieren tratamiento. En la presente revisión, se describen los distintos aspectos clínicos y terapéuticos de esta enfermedad, resaltando la evidencia respecto al estudio diagnóstico del compromiso extracutáneo.
Juvenile xanthogranuloma (JXG) is a rare benign disorder. It is the most common form of non-Langerhans cell histiocytosis (LCH)1,2 and specifically belongs to the C group (cutaneous and mucocutaneous histiocytoses). It results from a proliferation of dendritic cells that are positive for factor XIIIa, which is assumed to be the precursor of many dendritic non-LCH disorders.2,3 JXG was first described by Adamson4 in 1905 in a report of congenital xanthoma multiplex in a 2-year-old boy.4 Seven years later, McDonagh described 5 very similar cases,5 but it was not until the 1950s that Helwig and Hackney,6 in a series of 140 cases, first used the term juvenile xanthogranuloma.6
JXG usually manifests as erythematous yellowish papules or nodules typically located on the head and neck. Extracutaneous involvement is very rare and most cases affect the eyes.2,7,8
In this article, we described the main epidemiologic, etiologic, pathogenic, clinical, diagnostic, and treatment aspects of JXG, with a focus on the evidence regarding assessment of extracutaneous involvement. We also provide an algorithm for both the assessment and management of extracutaneous involvement.
EpidemiologyThe true incidence of JXG is unknown. The estimated prevalence is about 1 case per million children, but it is thought to be much higher, as small isolated lesions may be underdiagnosed.9 In the Kiel Pediatric Tumor Register, which collected data covering a period of 35 years, JXG accounted for 129 (1.5%) of 24 600 pediatric tumors.10
JXG is mainly a pediatric disease, with adult-onset cases accounting for just 10% of cases. The lesions appear by the age of 7 months in 64% of patients and within a year in 85%. Up to a third of lesions may be congenital.2,9 A younger age at presentation has been linked to more diffuse forms of the disease. JXG is more common in males (male to female ratio, 1.1-7:1), but no differences in prevalence rates between men and women have been observed for adult-onset JXG1,10 (Table 1).
Epidemiological, Clinical, and Histologic Characteristics of Juvenile Xanthogranuloma.
| Epidemiology | -True incidence unknown-Prevalence of 1 case per million children-85% of lesions develop within first year of life-One-third of lesions may be congenital-Male to female ratio: 1-7:1 |
|---|---|
| Clinical presentation | -Well-defined red and yellow or orange papule or nodule with a smooth surface and firm consistency-Solitary lesion in 80% of cases-Most common location: head and neck-Atypical forms: generalized, lichenoid, giant, mixed, plaque, subcutaneous, annular, muscular, agminated, cutaneous horn |
| Dermoscopy | -Setting sun pattern: central yellow-orangish area with pale yellow areas and an erythematous halo-Peripheral linear telangiectasias may be observed |
| Histology | -Dense, well-demarcated lymphohistiocytic infiltrate, with giant cells (Touton, foreign body, ground glass)-Positive staining for CD68, CD163, KiM1P, anti-FXIIIa, vimentin, and anti-CD4 and negative staining for S-100, CD1a, and CD207 |
| Extracutaneous manifestations | -Most common location: eyes-Less common locations: spleen, lungs, and central nervous system-Rare locations: heart, bone marrow, gastrointestinal tract, pancreas, bones, retroperitoneum, adrenal glands, testicles, and kidneys-May be present in up to 10% of patients with neurofibromatosis type 1 |
The pathogenesis of JXG remains uncertain. Traditionally, it has been considered to be a non-neoplastic process characterized by an abnormal response to a nonspecific injury, such as trauma or viral infection.11 This view, however, contrasts with more recent evidence showing certain genetic associations.12,13
Why histiocytes become increasingly lipid-laden in the absence of hyperlipidemia remains unclear. Low-density lipoprotein uptake and increased cholesterol synthesis within macrophages have been described in adults with JXG.14
Recent whole-exome sequencing studies have pointed to a possible role for pathological extracellular signal-regulated kinase (ERK) activation. It has been proposed that activation of the ERK pathway in the germinal stage of development or in bone marrow stem cells might lead to aggressive multiorgan disease, while activation at a later stage of cell differentiation might result in disease affecting a single or just a few organs and unifocal lesions, as occurs in JXG.12 Chakraborty et al.13 identified 17 somatic mutations in 4 JXG lesions (a median of 4 mutations per lesion). Although they did not identify any BRAF-V600E mutations (associated with LCH), they did detect a PI3KCD mutation in one patient and a germline mutation in the gene coding for neurofibromin in another with neurofibromatosis type 1 (NF-1).
Clinical Presentation- a
Cutaneous Manifestations
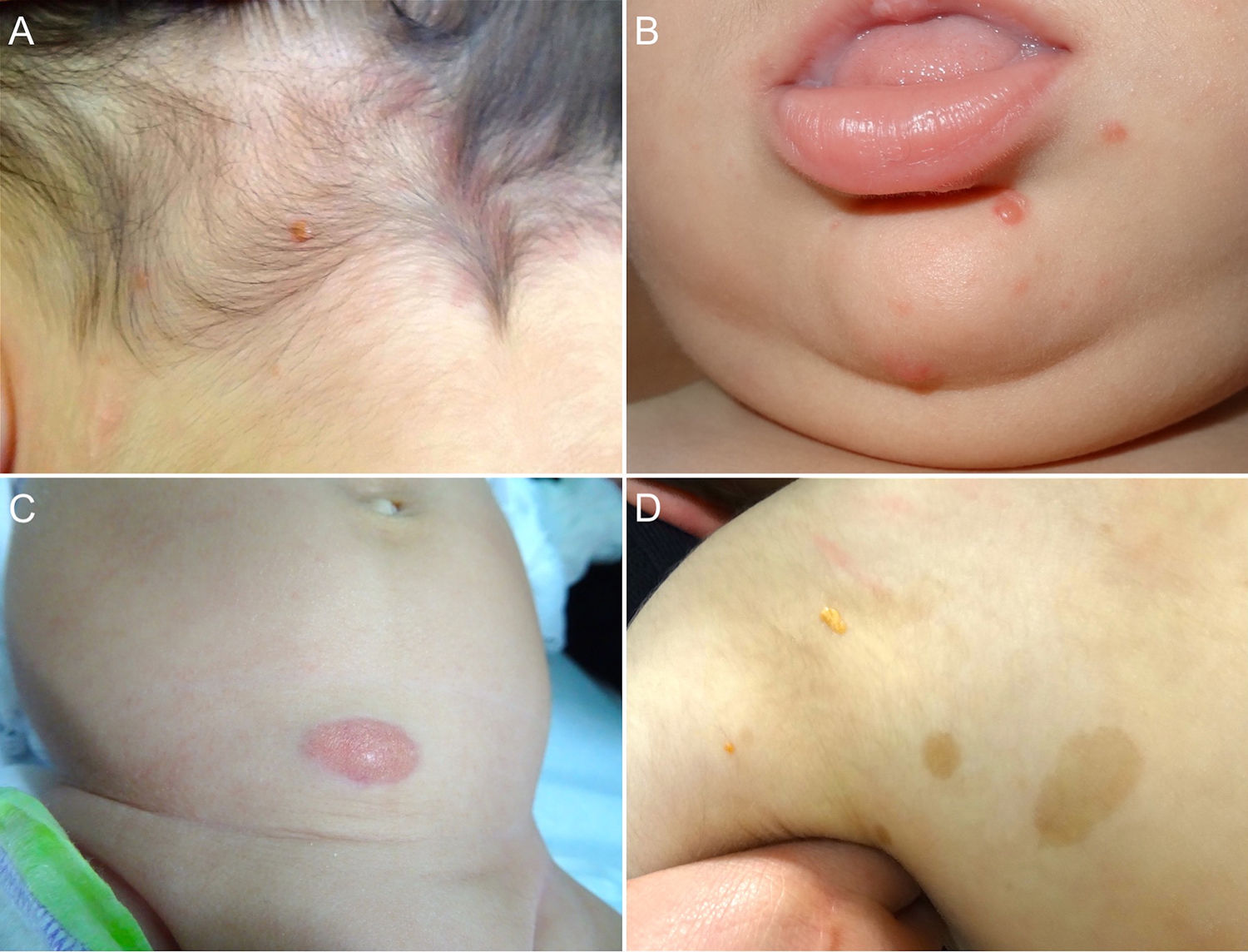
Cutaneous lesions are the most common manifestation of JXG. Two classification systems based on lesion size have been described. In the first system, Caputo et al.15 distinguishes between 3 main types: small nodular or papular (2−5 mm), large nodular (5−20 mm), and giant xanthogranuloma (> 20 mm). In the second system, devised by Gianotti et al.,16 the distinction is between micronodular (< 10 mm, usually multiple lesions) and macronodular (10−20 mm) (Table 2). Although clinically JXG is characterized by a wide spectrum of lesions in terms of shape, color, and number, up to 80% of lesions are solitary. Multiple JXGs are more common in children, with numbers ranging from 2 or 3 to over 100.3,17 The characteristic JXG lesion is a well-demarcated papule or nodule with a smooth surface. In its initial stages, it has a typically pink-reddish color, but it progressively acquires a characteristic yellow, orange, or brownish color. The lesions have a firm consistency and some display superficial telangiectasias. With the exception of certain locations or very large lesions, JXG is usually asymptomatic. The most common location is the head and neck, followed by the trunk and limbs1,18,19 (Fig. 1A, B) (Table 1).
 lesion in the occipital region. B, Eight-month-old boy with multiple congenital JXG lesions on his chin. C, Four-month-old girl with a giant congenital JXG lesion on her abdomen. D, JXG associated with café au lait spots in a 4-year-old boy with neurofibromatosis type 1.")
A, One-year-old boy with a solitary juvenile xanthogranuloma (JXG) lesion in the occipital region. B, Eight-month-old boy with multiple congenital JXG lesions on his chin. C, Four-month-old girl with a giant congenital JXG lesion on her abdomen. D, JXG associated with café au lait spots in a 4-year-old boy with neurofibromatosis type 1.
Atypical variants of JXG include generalized, lichenoid, giant (>2 cm in diameter), mixed, plaque, subcutaneous, annular, muscular, and clustered forms, as well as a cutaneous horn.20,21
Congenital JXG appears to have an even more varied clinical spectrum than the classic variant. Congenital lesions are typically large solitary tumors with an atypical morphology, and there have also been reports of deep subcutaneous lesions, infiltrative plaques, exophytic tumors, and agminated plaques, among others22 (Fig. 1C). The 2 most common complications of congenital JXG are atrophic scarring and ulceration, with the latter appearing to be more common in large exophytic lesions.22,23
- b
Extracutaneous Manifestations
Although JXG lesions are mostly limited to the skin, multiple cases of extracutaneous lesions involving a single site or various organs (mostly the eyes) have been reported. A recent retrospective study of 2949 pediatric cases described in the literature found that just 0.24% had ocular involvement. The authors therefore concluded that referral of a child with JXG without ocular or visual manifestations for an eye examination would have a very low yield.24 Other authors have found that approximately 40% of patients with ocular JXG had a history of skin lesions, which can appear between 8 and 10 months before or after the ocular manifestations.25
Ocular JXG is nearly always unilateral and generally affects the iris, although the orbit, optic nerve, choroid, and conjunctiva may also be affected. The most common presenting symptom is a unilateral red eye, followed by a tumor in the iris or conjunctiva. Ocular lesions, unlike cutaneous lesions, do not resolve spontaneously and can cause hyphema, glaucoma, and/or loss of vision.17,25,26 Three risk factors have been proposed: 1) micronodular JXG, 2) multiple skin lesions, and 3) early onset (≤ 2 years).16,17 We would therefore recommend referring patients with 2 or more skin lesions or early-onset lesions for an eye examination.
Systemic JXG is a rare histiocytic disorder that usually manifests as multiple cutaneous and/or subcutaneous nodules and involvement of 2 or more visceral organs.27 Just 61 cases of systemic JXG have been reported to date. The most frequently affected organs are the liver, spleen, lungs, eyes, and central nervous system, but there have also been anecdotal cases involving the heart, bone marrow, gastrointestinal tract, pancreas, bones, retroperitoneum, adrenal glands, testicles, and kidneys.1,28–31 Systemic JXG occurs at a younger age (mean, 5.5 months) than classical JXG and the male to female ratio is 1.4:1. Multiple skin lesions, postulated as a risk factor for systemic involvement, have been observed in 67% of patients with systemic JXG, with a latency period ranging from months to several years.24,29 Nondetection of extracutaneous involvement in a patient with multiple JXG lesions, therefore, does not rule out subsequent development of systemic JXG. It would therefore seem reasonable to schedule follow-up visits to check for systemic disease.
A thorough physical examination should be performed at diagnosis to assess the presence of systemic involvement.32 The clinical manifestations will vary according to the affected organ, with severity ranging from asymptomatic to fatal. Involvement of the central nervous system and liver has been linked to significant morbidity and mortality.28,29
JXG lesions occur in approximately 5% to 10% of patients with NF-133 (Fig. 1D). Prevalence is influenced by the child's age, as JXG lesions tend to resolve around the age at which NF-1 becomes clinically evident. Although the link between JXG and NF-1 remains unclear, several authors have highlighted the importance of checking for NF-1 stigmata in children with multiple JXG lesions.34 The description of a triple association involving JXG, NF-1, and juvenile myelomonocytic leukemia (JML) has sparked frequent debate. In 1995, Zvulunov et al.35 concluded that this triple association was 30 to 40 times more common than would be expected by chance and estimated that children with NF-1 and JXG were 20 to 32 times more likely to develop JML than those with NF-1 alone. Nonetheless, the study had some methodological and statistical limitations that may have resulted in overestimation. Two subsequent retrospective studies did not find an increased incidence of JML in patients with JXG and NF-1.36,37 The lack of association was further supported by the results of a more recent retrospective case-control study of 739 patients with NF-1 spanning 20 years. Fourteen of the 14 patients (1.9%) developed malignancy, and of these, 4 (28.5%) developed JXG. The difference with the control group (6/29 patients, 21%) was nonsignficant.33 Demonstration of a lack of association between JXG and malignancy in patients with NF-1 would be good news for both doctors and parents. Because JXG and hematologic/solid tumors are associated with NF-1, their coexistence may be a pure coincidence.33,38 Patients with NF-1 should be screened for malignancy (in particular, malignant peripheral nerve sheath tumor), regardless of whether or not they have JXG.39
Diagnosis- a
Clinical Diagnosis
The diagnosis of JXG is essentially clinical and is guided by the history, progression, and morphology of lesions. In some cases, diagnosis needs to be confirmed by histology or immunohistochemistry.2,3
- b
Dermoscopy
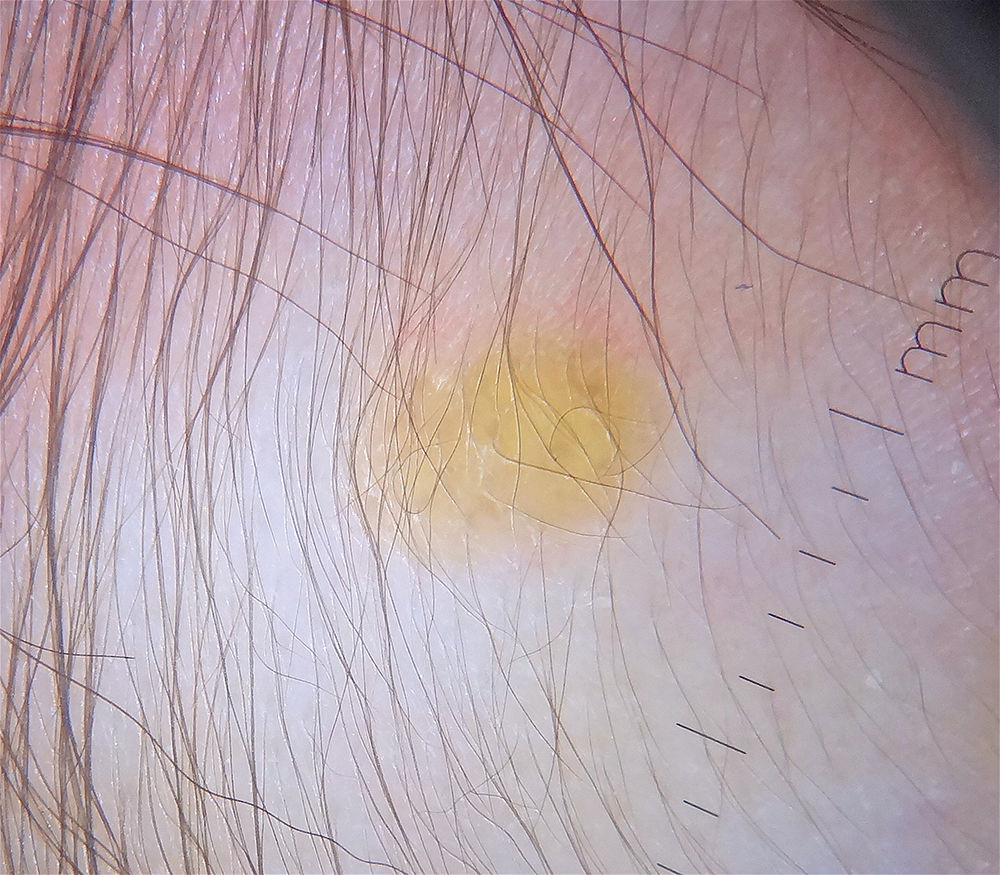
Dermoscopy is a useful noninvasive technique for the clinical diagnosis of JXG. One group of authors described a setting sun pattern, characterized by a central yellow-orange area that may show “clouds” of pale yellow deposits and an erythematous halo. This pattern can be observed in lesions of all stages.40 (Fig. 2). Peripheral linear telangiectasias and other nonspecific characteristics such as a discrete pigment network, whitish lines, and thin arborizing vessels have also been described.40,41
- c
Histology
.")
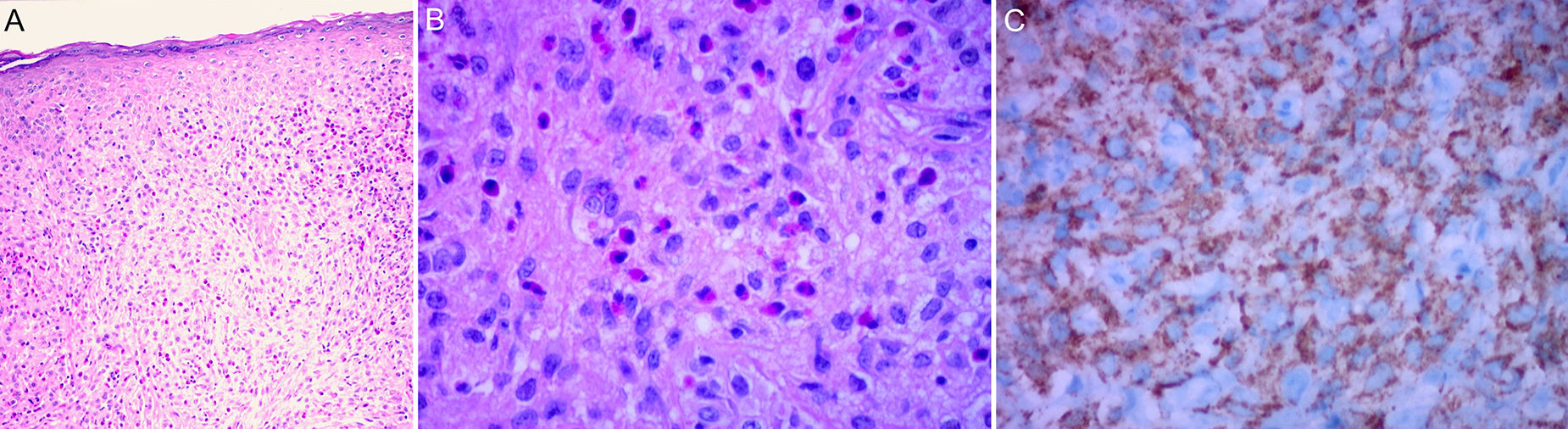
The classic histologic features of JXG include a dense, well-demarcated, noncapsulated, sheetlike histiocytic infiltrate in the dermis and upper subcutaneous tissue, with sparing of the epidermis and skin appendages. Five main cell types, present in varying proportions (from monomorphous to mixed variants), have been described in JXG infiltrates: vacuolated, xanthomatous, spindle-shaped, scalloped, and oncocytic. Variants of giant cells include nonspecific, foreign body, Touton, and ground-glass11 (Fig. 3A and B). Histologic findings vary according to lesion stage, with early-stage lesions showing a monomorphous infiltrate with lipid-free macrophages that can occupy most of the dermis and later-stage lesions with abundant vacuolated foamy macrophages and Touton-type multinucleated giant cells, particularly in the superficial dermis.1,11
. C, Immunohistochemistry. CD68+ marker.")
Touton cells, a typical finding in JXG, are characterized by a ring of nuclei around a cytoplasm with high lipid content. It has been estimated, however, that they are absent in around 15% of cases. Touton cells have been traditionally described as being pathognomonic for JXG, but they can also be found in other xanthomatous and histiocytic lesions (Erdheim-Chester disease and necrobiotic xanthogranuloma) and even dermatofibromas.1,2
In the immunohistochemical study, JXG lesions are positive for macrophage markers, such as CD68, CD163, KiM1P, anti-FXIIIa, vimentin, and anti-CD4, and generally negative for S-100, CD1a, and CD207 (antilangerin), which is specific to Langerhans cells11,42 (Fig. 3C).
Electron microscopy reveals discrete histiocytes with multiple intracytoplasmic lipid droplets. Other nonspecific intracytoplasmic findings include dense, worm-like, and popcorn bodies. The characteristic Birbeck granules seen in LCH are absent.43
Fine-needle aspiration cytology has also been described as a useful diagnostic technique for JXG. Cytologic smears show finely vacuolated, lipid-laden, mononucleated cells with reniform or oval nuclei against a mixed cell inflammatory infiltrate containing a variable proportion of lymphocytes, neutrophils, and eosinophils, along with Touton or foreign body-like giant cells. Nonetheless, a high index of suspicion on the part of the cytopathologist is required.44
- d
Doppler Ultrasound
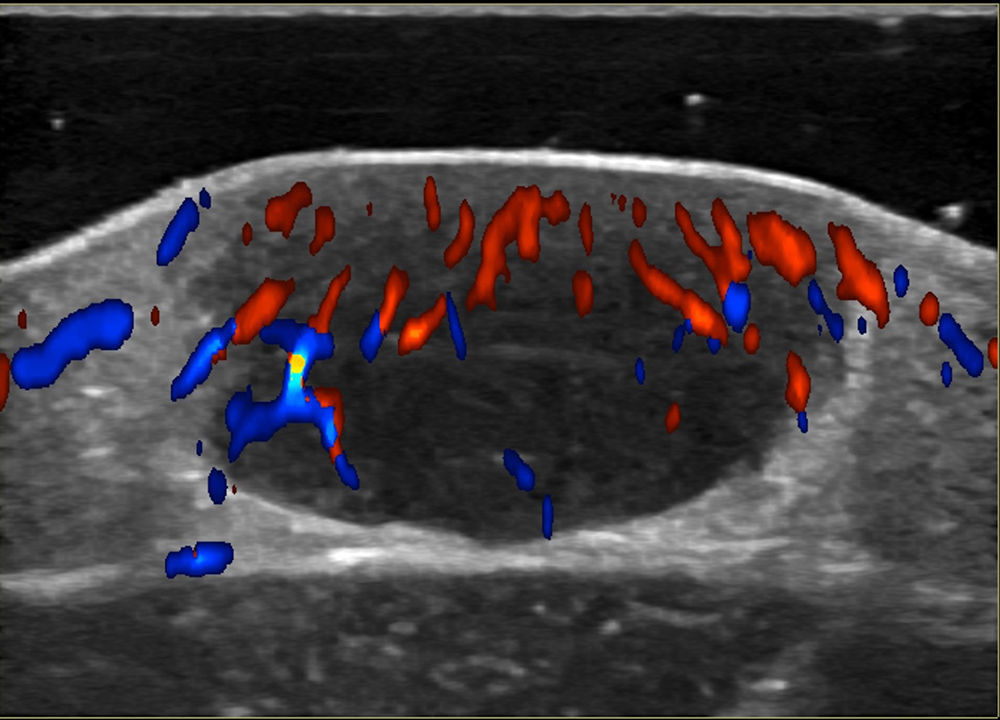
Noninvasive techniques such as Doppler ultrasound can be useful alternatives to histology for assessing and monitoring lesions. Ultrasound findings include a well-demarcated hypoechoic nodule at the dermal-epidermal junction or in the dermis without posterior enhancement or lateral shadowing. In some cases, color Doppler ultrasound shows vascularization, with thin, slow-flow arteries within the lesion (Fig. 4).45,46
Assessment of Extracutaneous Involvement
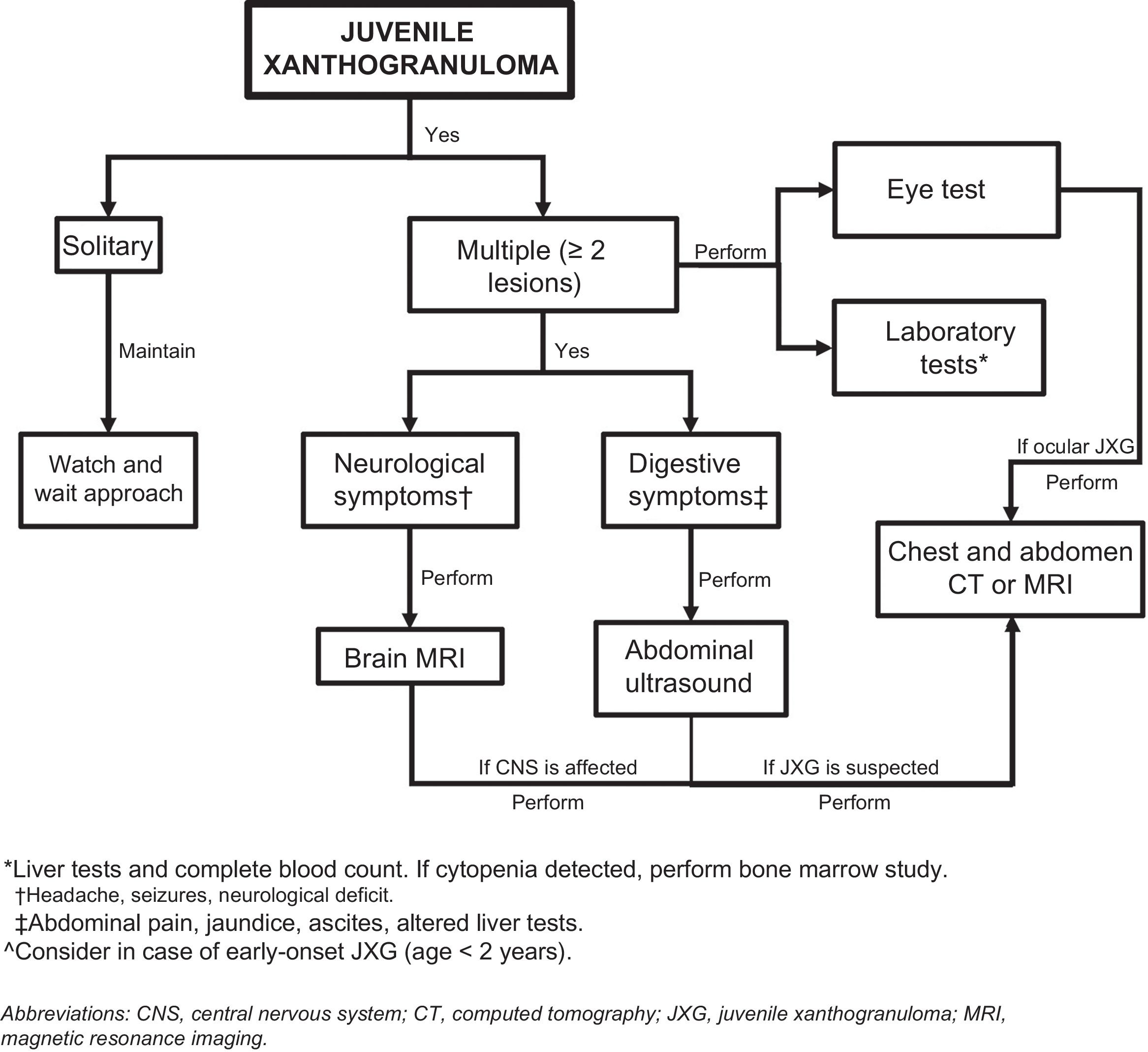
Considering that extracutaneous involvement is rare, consensus is lacking on the value of additional tests in the absence of suggestive signs or symptoms.32 Depending on the patient’s history, Höck et al.11 suggests that the diagnostic work-up could include, in addition to a full skin examination, a laboratory work-up, ultrasound examination of lymph nodes and the abdomen, a chest X-ray, a skeletal X-ray or a radionuclide bone scan, computed tomography (CT) or brain magnetic resonance imaging (MRI), and an eye examination. Based on the case of a 2-month-old infant with skin, lung, and liver involvement, Meyer et al.47 suggested that all children with 2 or more cutaneous JXG lesions should undergo screening for systemic JXG, including CT or MRI of the brain and chest/abdomen. Doppler echocardiography should be performed if heart involvement is suspected. The laboratory work-up should include a complete blood count (followed by bone marrow aspiration/biopsy if cytopenia is detected) and a complete metabolic profile including kidney and liver function.
Considering the few cases of systemic JXG that have been reported and the benign, self-limiting nature of cutaneous and extracutaneous lesions, it would seem reasonable to base the diagnostic work-up on clinical manifestations and physical findings and consideration of the most frequently involved organs (Fig. 5). New studies, however, are needed to provide better evidence to inform more accurate recommendations.
Differential Diagnosis
The differential diagnosis of JXG depends on clinical presentation and lesion location and includes benign and malignant entities.
Considering its prognostic and therapeutic implications, LCH is one of the main entities that should be contemplated in the differential diagnosis, particularly in patients with multiple skin lesions and systemic involvement. Histologic examination can confirm a diagnosis of LCH, as it will show nuclei with a coffee-bean appearance. LCH lesions will also stain for S-100, CD1a, and CD207 in the immunohistochemical study. There have several reports of JXG in patients with LCH, with lesions appearing within under 5 years. The association might be due to a biological association between the 2 entities or, as most cases have involved patients who had received or were receiving chemotherapy at the time of JXG onset, an unknown mechanism induced by chemotherapy whereby LCH is transformed to JXG.48,49
Certain forms of non-LCH such as benign cephalic histiocytosis, necrobiotic xanthogranuloma, and multicentric reticulohistiocytosis should also be contemplated in the differential diagnosis.2,27,50 Small nodular JXG may be difficult to distinguish from benign cephalic histiocytosis, although the latter may display certain characteristics that aid its detection, such as symmetrically distributed lesions, cephalocaudal progression, palmoplantar involvement (albeit rare), nasal and scalp involvement, and absence of systemic involvement.51
Other lesions that can imitate JXG include pediatric cellular neurothekeoma, tuberous xanthomas, molluscum contagiosum, maculopapular cutaneous mastocytosis, mastocytomas, hemangiomas, neurofibromas, dermatofibromas, Spitz nevi, histiocytomas, congenital reticulohistiocytosis, sarcoidosis, and xanthomas.9,52–54
TreatmentJXG does not typically require treatment as it generally follows a benign course, with lesions resolving spontaneously within approximately 3 to 6 years.1,2 However, in certain cases of adult-onset xanthogranuloma, lesions tend to persist and are less likely to resolve without treatment.55
Solitary lesions that cause functional impairment or psychological distress because of their location (particularly among parents) can be surgically removed or treated using other ablative methods, such as carbon dioxide laser therapy. A watch and wait approach is the first-line option for the vast majority of asymptomatic lesions.11
Topical and intralesional corticoids are usually used to treat ocular JXG, while systemic corticosteroids or surgery may be needed to treat rapidly progressive lesions or complications, such as glaucoma or hyphema; there have also been isolated reports of good response to radiation therapy and chemotherapy.25,29
Treatment of systemic JXG depends on the degree of visceral dysfunction caused by these benign lesions. Considering that most cases will follow a self-limiting course, a watch and wait approach is generally recommendable. In other words, treatment should be initiated when JXG starts to interfere with vital organ function. Options described include surgical resection, radiation therapy and/or chemotherapy with prednisone and vinblastine regimens used in LCH, or, for refractory cases, cytarabine and 2-chlorodeoxyadenosine.3,29,53 Nonetheless, given the scarcity of cases described in the literature and the consequent absence of treatment guidelines, individualized, multidisciplinary treatment is necessary.
ConclusionsJXG is a rare disorder with a wide spectrum of clinical manifestations that requires a comprehensive assessment to detect extracutaneous involvement. The evidence in such cases is limited and a diagnostic workup guided by clinical findings would appear to be a reasonable strategy. Treatment tends to involve watchful waiting, although chemotherapy may be required for systemic cases. Studies evaluating the merits of different regimens are needed to optimize their use in this multifaceted disease.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Hernández-San Martín MJ, Vargas-Mora P, Aranibar L. Xantogranuloma juvenil: una entidad con amplio espectro clínico. Actas Dermosifiliogr. 2020. https://doi.org/10.1016/j.ad.2020.07.004



