












This series of 2 articles on dermatopathologic diagnoses reviews conditions in which granulomas form. Part 1 clarifies concepts, discusses the presentation of different types of granulomas and giant cells, and considers a large variety of noninfectious diseases. Some granulomatous diseases have a metabolic origin, as in necrobiosis lipoidica. Others, such as granulomatous mycosis fungoides, are related to lymphomas. Still others, such as rosacea, are so common that dermatologists see them nearly daily in clinical practice.
En esta serie de dos artículos realizamos una revisión de las principales entidades dermatopatológicas que cursan con granulomas. Esta primera parte se ha centrado en la aclaración de conceptos, la presentación de los tipos de granulomas y de células gigantes, así como en entidades muy diversas de origen no infeccioso. Algunas de ellas de origen metabólico, como la necrobiosis lipoidica: otras relacionadas con linfomas, como la micosis fungoides granulomatosa; y otras tan extendidas que casi resultan un problema cotidiano en las consultas de dermatología, como la rosácea.
In dermatopathology, a granuloma is defined as a more or less tight aggregate of macrophages. Macrophages originate from bone marrow–derived monocytes, which are circulating mononuclear cells that infiltrate tissue and develop into cells with phagocytic capacity (macrophages or histiocytes).
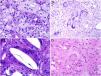
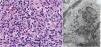
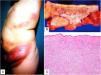
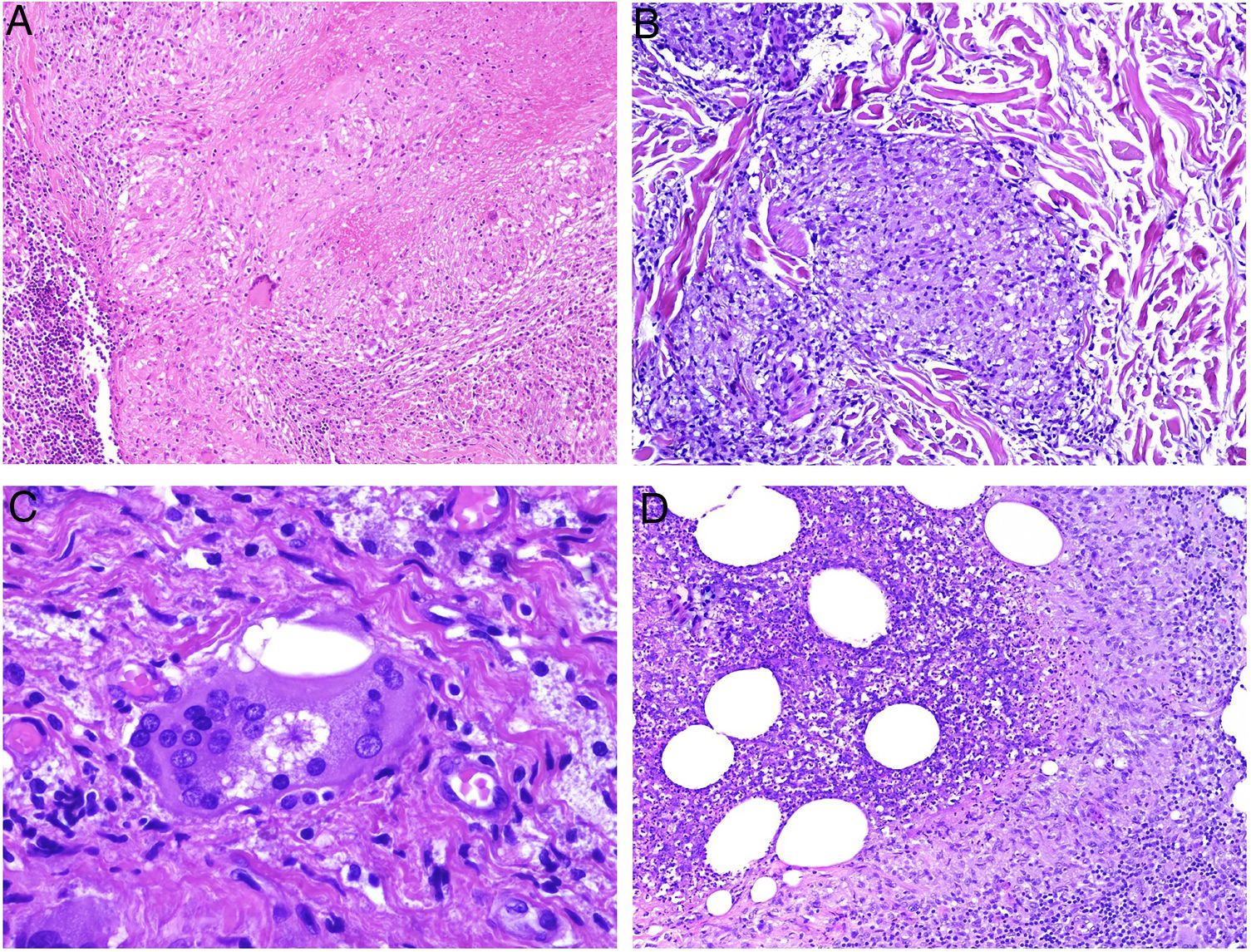
Histiocytes in granulomas tend to have abundant cytoplasm, and therefore resemble epithelial cells, hence the terms epithelioid histiocytes and epithelioid granulomas. Histiocytes are often located so close together that their borders are undistinguishable and they resemble a syncytium. Strictly speaking thus, scattered aggregates of macrophages are not considered granulomas. Xanthomatous histiocytes are histiocytes that acquire a foamy cytoplasm when they accumulate sufficient quantities of intracellular lipid (Fig. 1A).
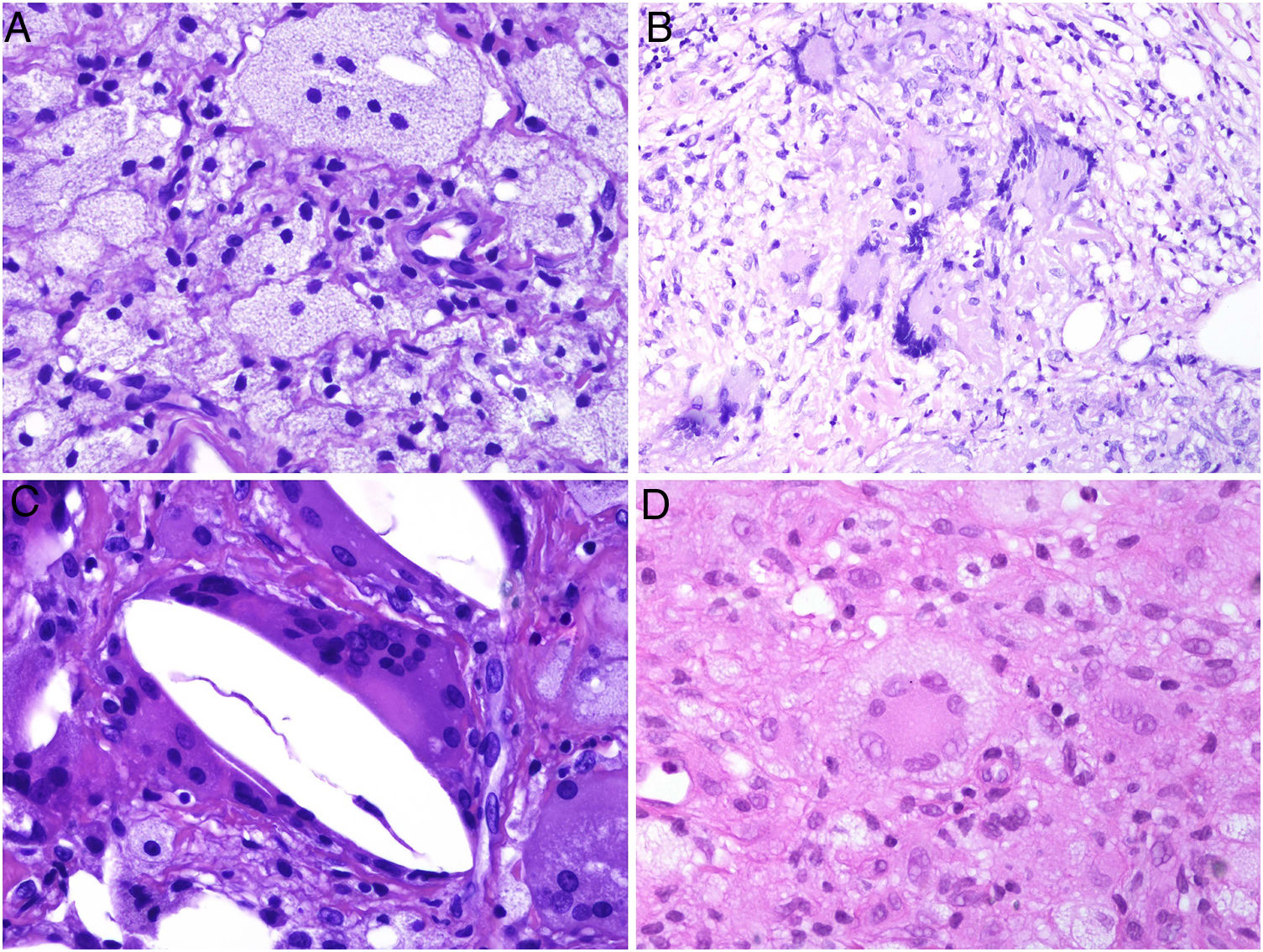
A, Xanthomatous cells. Histiocytes with high cytoplasmic lipid content, conferring them a multivesicular appearance (H&E, original magnification x400). B, Langhans cells. Multiple nuclei arranged towards the pole of the cell in a horseshoe pattern (H&E, original magnification x400). C, Foreign body giant cells. Two of these cells are encapsulating an empty acicular space corresponding to a cholesterol crystal. Note the multiple disordered nuclei in both cases (H&E, original magnification x400). D, Touton giant cell. Note the ring-like arrangement of nuclei. The cytoplasm within these rings is intensely eosinophilic, while the outer rim has a xanthomatous appearance (H&E, original magnification x400). H&E indicates hematoxylin-eosin.
In many granulomas, frustrated phagocytosis can give rise to large multinucleated histiocytes, known as multinucleated giant cells. Some variants have specific histopathologic characteristics and are more common in certain types of granulomas. It is important thus to be familiar with the different variants, described below:
- -
Langhans giant cells are cells with nuclei arranged towards the pole of the cell forming a horseshoe pattern; they have a dense eosinophilic cytoplasm (Fig. 1B).
- -
Foreign body giant cells have multiple (often > 10) dispersed nuclei. It is not uncommon for them to partly encapsulate the foreign body implant and even adapt to its morphology (Fig. 1C).
- -
Touton giant cells are round cells with a ring-like arrangement of nuclei. A peripheral rim of cytoplasm often remains between the nuclei and plasma membrane; this has a foamy appearance, unlike the central cytoplasmic areas, which are densely eosinophilic (Fig. 1D). Touton giant cells are typically found in inflammatory reactions with high lipid content.
There are different forms of granulomas (Fig. 2) and each may display different types of giant cells. None of the granulomas are pathognomonic, but they are all preferentially associated with 1 or more entities.
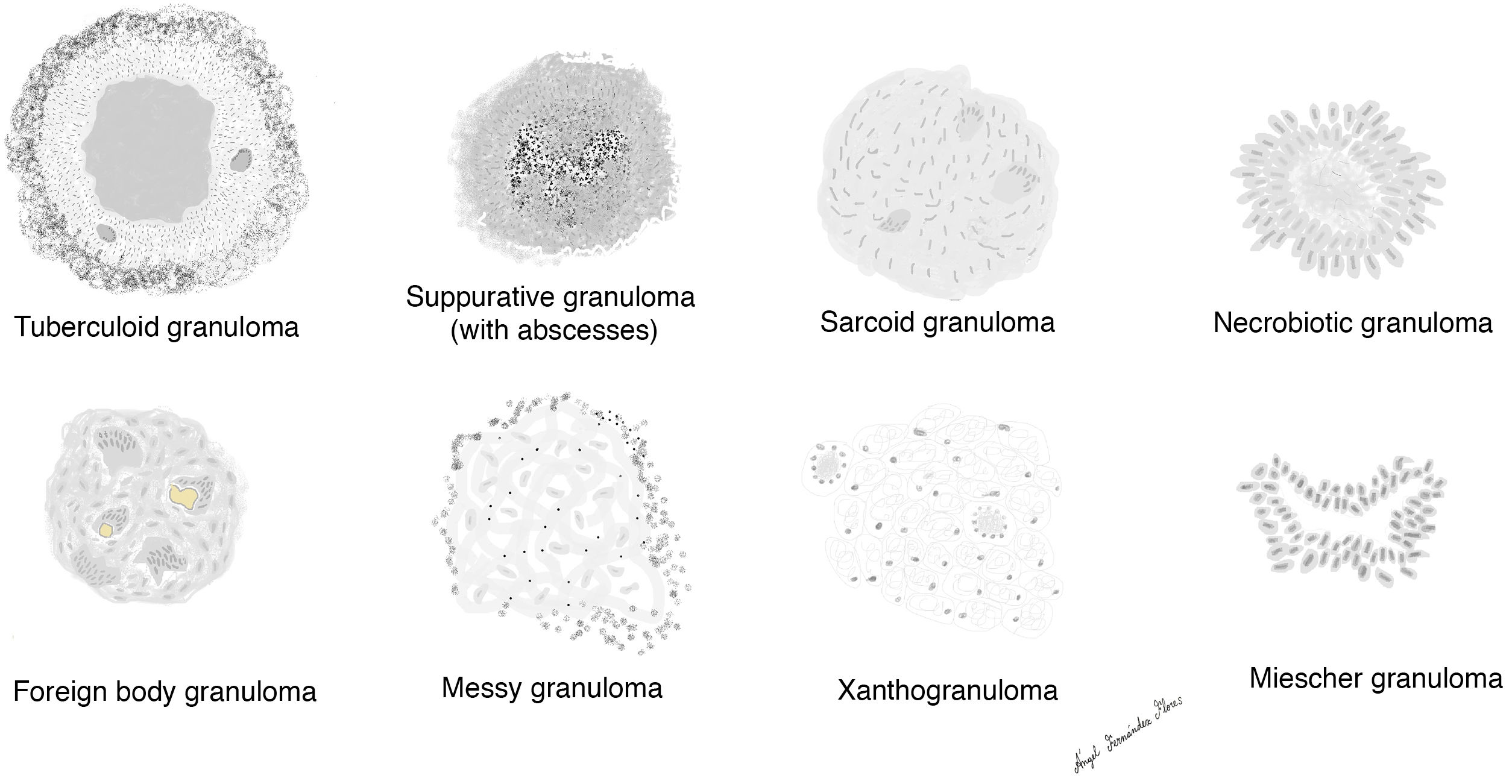
The main types of granulomas recognized to date are:
- -
Tuberculoid granulomas
- -
Sarcoid granulomas
- -
Suppurative granulomas (with abscesses)
- -
Foreign body granulomas
- -
Xanthogranulomas
- -
Messy granulomas
- -
Necrobiotic granulomas
- -
Miescher granulomas
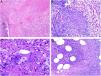
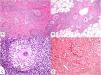
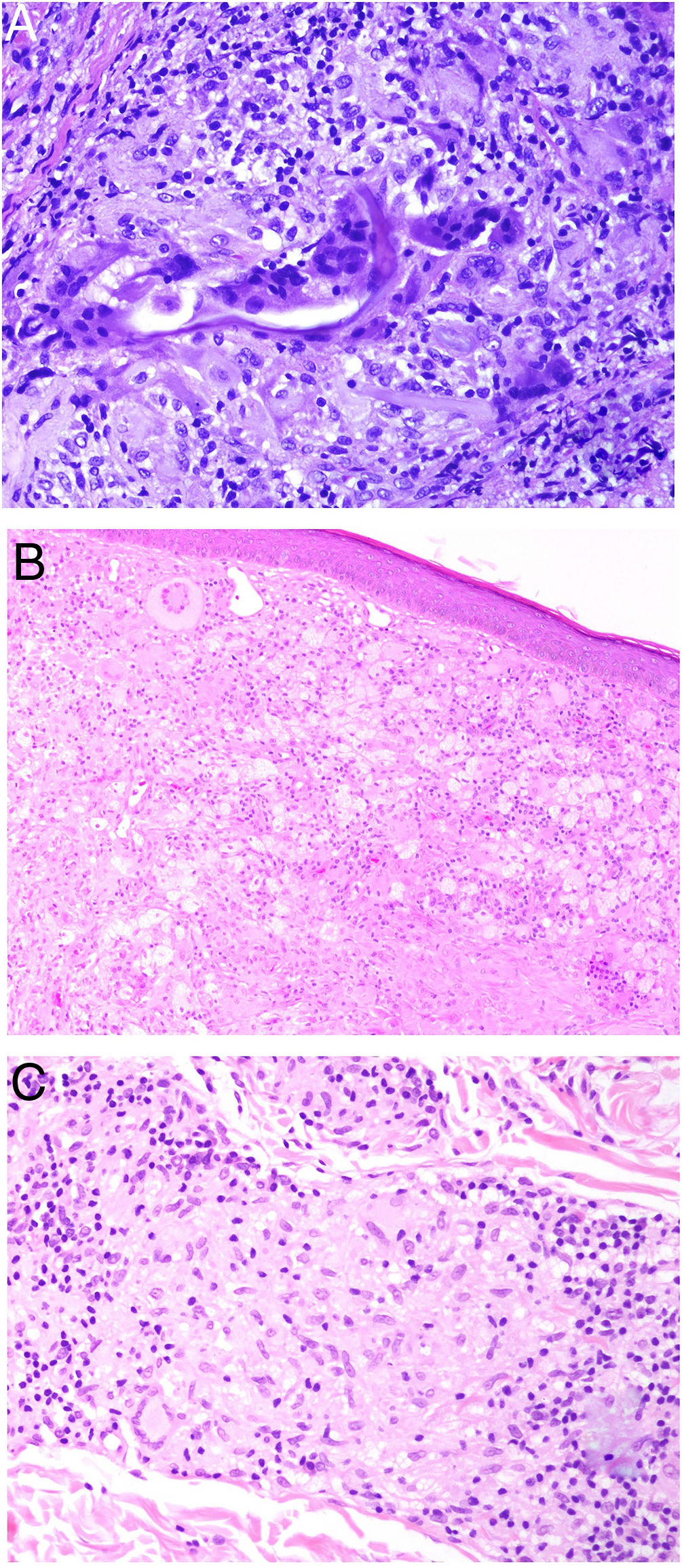
Tuberculoid granulomas are epithelioid granulomas that usually display central caseous necrosis, which is a form of coagulative tissue necrosis. As such, it is amorphous and practically homogeneous. The term caseous refers to the cottage cheese–like appearance of the dead tissue. Tuberculoid granulomas tend to have a prominent peripheral lymphocytic infiltrate that is sometimes compared to a crown (Fig. 3A). Langhans giant cells are common. Tuberculoid granulomas are the most common type of granuloma in tuberculosis.
A, Tuberculoid granuloma. Central areas of caseous necrosis surrounded by epithelioid histiocytes and an evident Langhans-type giant cell. Note the peripheral crown of lymphocytes (H&E, original magnification x100). B, Sarcoid granuloma. Non-necrotizing epithelioid granuloma without a prominent crown of lymphocytes (naked) (H&E, original magnification x100). C, Intracytoplasmic asteroid body with a star-like shape that is characteristic but not pathognomonic of sarcoid granuloma (H&E, original magnification x400). D, Suppurative granuloma (with abscesses) associated with atypical mycobacterial disease. Epithelioid histiocytes around a center of polymorphonuclear neutrophils (H&E, original magnification x100). H&E indicates hematoxylin-eosin.
Sarcoid granulomas, by contrast, do not have prominent central necrosis. When necrosis is present, it is discrete and fibrinoid. The fibrinoid nature of the necrosis is due to the deposition of fibrin alongside other components, such as antibodies. Sarcoid granulomas do not normally have a prominent peripheral inflammatory infiltrate, and are therefore described as naked (Fig. 3B). They are common in sarcoidosis. Giant cells with 2 types of intracytoplasmic inclusions are sometimes seen in sarcoidosis: star-like asteroid bodies (Fig. 3C) and Schaumann bodies, which are round, concentrically laminated calcified protein bodies. The inclusions are not pathognomonic of sarcoid granulomas.
Suppurative granulomas show abundant polymorphonuclear neutrophils in their center (Fig. 3D). They are most often seen in infections caused by fungi or atypical mycobacteria.
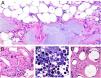
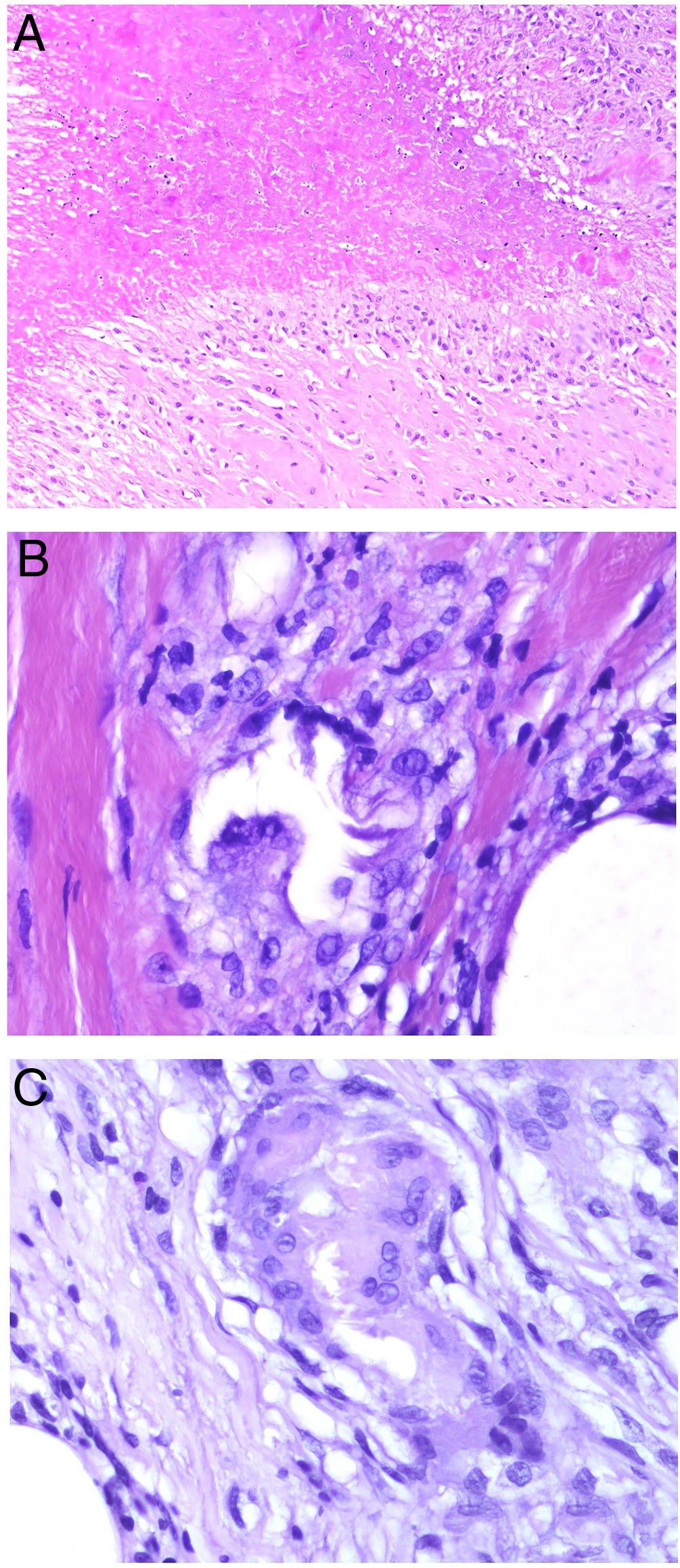
Foreign body granulomas are morphologically very varied, as they form in response to a wide range of foreign bodies. Giant cells often encapsulate and adapt their shape to the foreign particles they are attempting to engulf (Fig. 4A). Foreign body granulomas are composed of histiocytes and lymphocytes and do not have a consistent or predefined shape.
A, Foreign body granuloma. Numerous multinucleated giant cells with dispersed nuclei encapsulating the foreign body (H&E, original magnification x200). B, Xanthogranuloma. Abundant foamy cells alongside occasional Touton cells (H&E, original magnification x40). C, Messy granuloma associated with leishmania. Haphazardly arranged, widely separately histiocytes with clear cytoplasm (H&E, original magnification x100). H&E indicates hematoxylin-eosin.
Xanthogranulomas are mainly composed of histiocytes and giant cells with xanthomatous (foamy cytoplasmic) changes due to the accumulation of lipid content (Fig. 4B).
Messy granulomas are formed by loose, randomly arranged epithelioid histiocytes1 (Fig. 4C). The loose appearance is due to the abundant cytoplasm and glassy appearance of the histiocytes. They also appear to be separated by a hyaline substance that stains green with Masson trichrome stain. They are permeated by lymphocytes and tend to have a lymphocytic crown. In other words, they are not naked granulomas. Messy granulomas are a characteristic finding in leishmania.
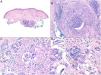
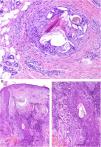
The term necrobiotic in necrobiotic granuloma suggests the presence of “dead” tissue in the center of the granuloma. This definition is controversial for a number of reasons. First, the central material is degenerated connective tissue (e.g., elastic or collagen fibers, mucin). Since connective tissue is not a cell, it cannot die, or be considered dead or living. What can—and in fact does—happen is that its properties degenerate or become altered. Second, a number of granulomas that could be considered true necrobiotic granulomas are not included in the category of necrobiotic granulomas (e.g., granulomas with central caseous necrosis that does originate from the death of cells). Necrobiotic granulomas include granuloma annulare, necrobiotic xanthogranuloma, rheumatoid nodules, and necrobiosis lipoidica. Because of the controversy surrounding the term necrobiotic, we believe it more appropriate to use the term extracellular degenerative granulomas. These granulomas are also known in the literature as palisaded granulomas, as the histiocytes surrounding the central area of degenerated tissue may follow a palisading pattern (Fig. 5A).
A, Necrobiotic (palisaded) granuloma corresponding to a rheumatoid nodule. Central necrobiotic material with palisading histiocytes (H&E, original magnification x40). B, Miescher granuloma associated with erythema nodosum. Central angulated space with peripheral histiocytes (H&E, original magnification x400). C, Maturing Miescher granuloma. With time, Miescher granulomas may come to be formed exclusively by multinucleated histiocytes (H&E, original magnification x400). H&E indicates hematoxylin-eosin.
Miescher granuloma is characterized by a collection of small histiocytes with pale cytoplasm surrounding a central cleft (Fig. 5B). As the granuloma matures, occasional peripheral giant cells may appear. The central cleft is clearly delimited by a cuticle. Giant cells can become the predominant or only component (Fig. 5C), but the central cleft and cuticle remain. The nature of these clefts is unknown. Miescher granuloma is quite specific to erythema nodosum.2
Dermatopathologic Entities With Granulomatous ReactionsIn this 2-part review, we describe the main dermatopathological entities characterized by a prominent granulomatous reaction.
Reactions to Cosmetic FillersDefinition. The dermal or subcutaneous injection of fillers to repair cosmetic defects can trigger adverse granulomatous reactions that are histologically reminiscent of the injected material.3
Clinical presentation. Clinical manifestations range from swelling or erythema to ulcers or fistulas, and can be early or late. Late manifestations are usually caused by nonbiodegradable products and are sometimes associated with sclerosis or displacement.3,4
Histopathology. Histopathologic patterns are highly variable and include foreign body granulomas, palisaded histiocytes surrounding collections of filler material, and interstitial, sarcoid, or suppurative granulomatous reactions. Eosinophils may be observed. The injected filler agent, or its silhouette, can often be discerned. Characteristic findings are Swiss cheese holes, translucent polygonal spheres or structures, blue spheres, spicules visible under polarized light, and aggregates of bluish material.5 Silicone is one of the most common filler agents involved and shows histiocytes with variably sized vacuoles containing translucent material (pseudolipoblasts) and frequent asteroid bodies. Hyaluronic acid, currently the most widely used filler agent, is usually viewed as mucinous lacunae (which stain with Alcian blue) interspersed with histiocytes; it can lead to fibrosis or encapsulation6 (Fig. 6). It is not uncommon to detect 2 materials. These may form part of the same compound or correspond to materials injected at different times (e.g., a second injection may have activated a material that had been inert for years).4
Granulomas associated with dermal fillers. A, The bluish material with a myxoid or mucinous appearance is hyaluronic acid. It is interspersed with histiocytes containing variably sized vacuoles (arrows) corresponding to previously injected silicone (H&E, original magnification x200). B, Granulomatous reaction containing polygonal acrylic methacrylate hydrogel structures (H&E, original magnification x400). C, Reaction to hyaluronic acid and dextran compound (H&E, original magnification x400). D, Polymethylmethacrylate spheres in bovine collagen (H&E, original magnification x400). H&E indicates hematoxylin-eosin. H&E indicates hematoxylin-eosin.
Xanthelasma-like histiocytic reactions to local filler injections are common.7 However, they are considered to be the result of local alterations of low-density lipoprotein metabolism more than a direct foreign body response.
Granulomas Due to Noncosmetic ProductsDefinition. Any object introduced into the body from outside is capable of acting as a foreign body, setting the immune system into action and triggering an inflammatory response. Foreign bodies include biologic material (e.g., hair and nail keratin, thorns or cactus spines, parts of jellyfish, coral, or sea urchins) and inorganic matter (e.g., tattoo ink, sutures, silicone, talc).8
Clinical presentation. The clinical manifestations of cutaneous foreign body reactions are highly variable; they are influenced by the type of foreign body and the inflammatory phase (acute or chronic). Most cases present as red to brown ulcerated or nonulcerated papules, nodules, or plaques that subsequently harden and become fibrotic. Other manifestations include pseudofolliculitis (reaction to beard hair follicles or shaven skin), abscesses, sinus tracts, and even sarcoid-like granulomatous lesions (especially in the case of tattoo ink).8
Histopathology. Foreign body implantation triggers an acute neutrophilic inflammatory reaction followed by a chronic inflammatory phase. Although different patterns of chronic tissue reactions have been described (e.g., lichenoid, eczematous, pseudolymphomatous), granulomatous reactions are the most common.8
Granulomatous inflammatory reactions (Fig. 7A,B) are usually characterized by macrophages, which tend to grow, forming epithelioid histiocytes, or fuse, forming multinucleated giant foreign body–type cells (Figs. 7C and D). T cells and fibroblasts are usually present.8 There have also been descriptions of allergic-type granulomas with epithelioid histiocytes and a lower density of Langhans-type giant cells.9
Granulomatous reaction to suture material. A, Panoramic view of surgical specimen showing 2 deep granulomas (H&E, original magnification x40). B, Detail showing a mixed inflammatory infiltrate with lymphocytes, macrophages, epithelioid histiocytes, and several multinucleated foreign giant cells around suture filaments (H&E, original magnification x200). C) Multinucleated foreign body giant cells may sometimes be the predominant cell type (H&E, original magnification x400). D, Mutinucleated giant cells digesting suture fibers (H&E, original magnification x1000). H&E indicates hematoxylin-eosin.
Histology can also help identify the type of foreign body involved. Polarized light microscopy, for example, shows positive birefringence for certain materials, such as silicone, talc, keratin, thorns, sutures, and arthropod parts. Periodic–acid Schiff (PAS) staining, in turn, is positive for wood splinters, thorns, and starch.8
Aluminum GranulomasDefinition. Vaccines containing aluminum salts for antigen adsorption can produce aluminum granulomas, which are persistent local reactions in the form of pruritic injection-site nodules. These reactions are rare. According to prospective studies, they have an estimated incidence of 0.83% in vaccinated children and may be slightly more common in adults undergoing allergy desensitization.10 Aluminum granulomas are associated with contact dermatitis to aluminum hydroxide in over 80% of cases.11
Clinical presentation. The lesions consist of pruritic subcutaneous nodules, often accompanied by epidermal changes caused by scratching, such as hypertrichosis, lichenification, excoriations, and erythema12 (Fig. 8A). They can occur weeks or even months after vaccination and can last for years if untreated.
Aluminum granuloma. A, Subcutaneous nodule with lichenification caused by scratching at vaccination site in a patient undergoing allergy desensitization. B, Infiltrate mainly occupying the hypodermis (H&E, original magnification x20). Granulomas consisting of lymphocytes, histiocytes, and eosinophils (H&E, original magnification x200). H&E indicates hematoxylin-eosin.
Histopathology. Because aluminum-based vaccines are injected subcutaneously, the main histopathologic findings associated with aluminum granulomas are located in the subcutaneous tissue (Fig. 8B). Three patterns have been described,13 the most common being lobular panniculitis, with lymphocytes, plasmocytes, histiocytes, and eosinophils. The second most common pattern is B-cell pseudolymphoma, consisting of lymphoid follicles with well-developed germinal centers separated by a granulomatous histiocytic infiltrate. The third most common pattern is that of a necrotizing granuloma in the subcutaneous tissue, with lymphocytes, plasmocytes, and eosinophils (Fig. 8C). The key to diagnosis in all cases is the observation of histiocytes with an abundant, coarse, violaceous cytoplasm (Fig. 9A), which corresponds to intracellular deposits of aluminum salts. The diagnosis can be confirmed by electron microscopy (Fig. 9B), although Epstein-Barr virus–encoded small RNA in situ hybridization may also have diagnostic potential, as a recent study showed positive staining of granular histiocytes.14
Necrobiosis Lipoidica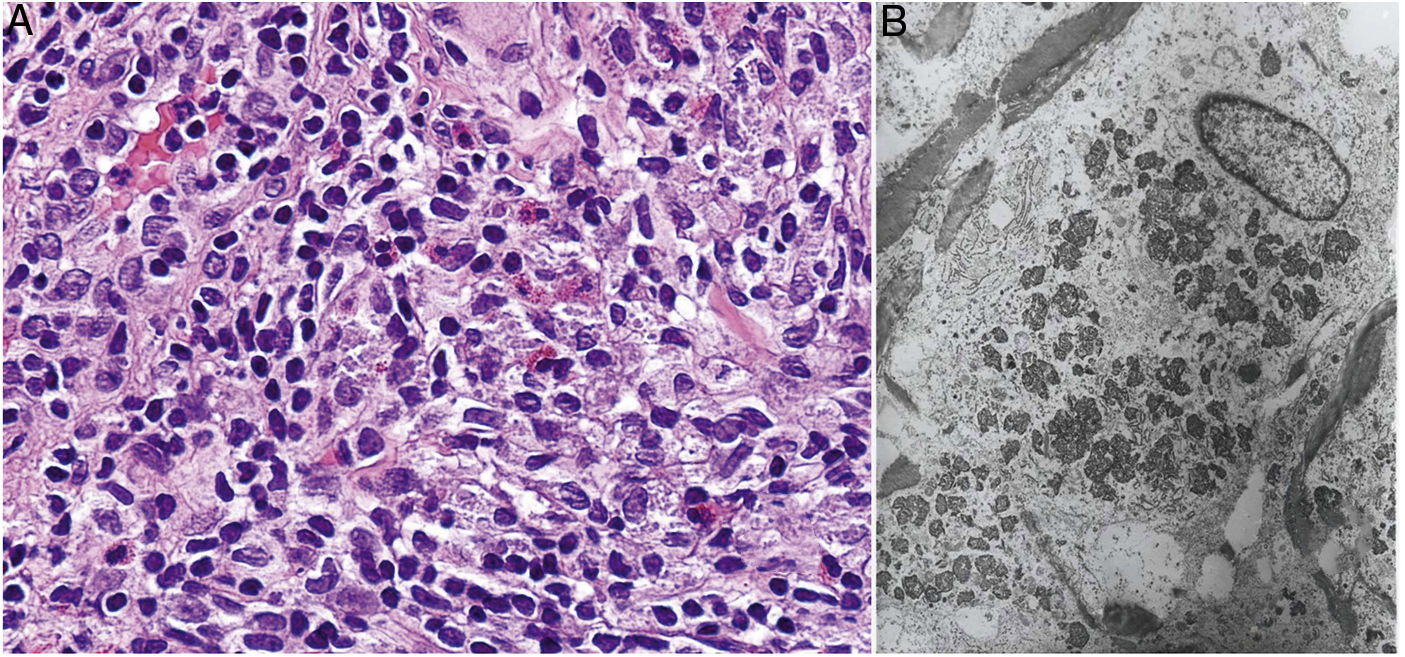
Definition. Necrobiosis lipoidica (NL) is a chronic idiopathic granulomatous disease that has traditionally been associated with diabetes mellitus (DM). Nonetheless, it has been estimated that less than 1% of patients with DM develop NL, and the percentage of diabetic patients in NL series is also highly variable.15–18 The origin and pathogenesis of NL have not yet been established.
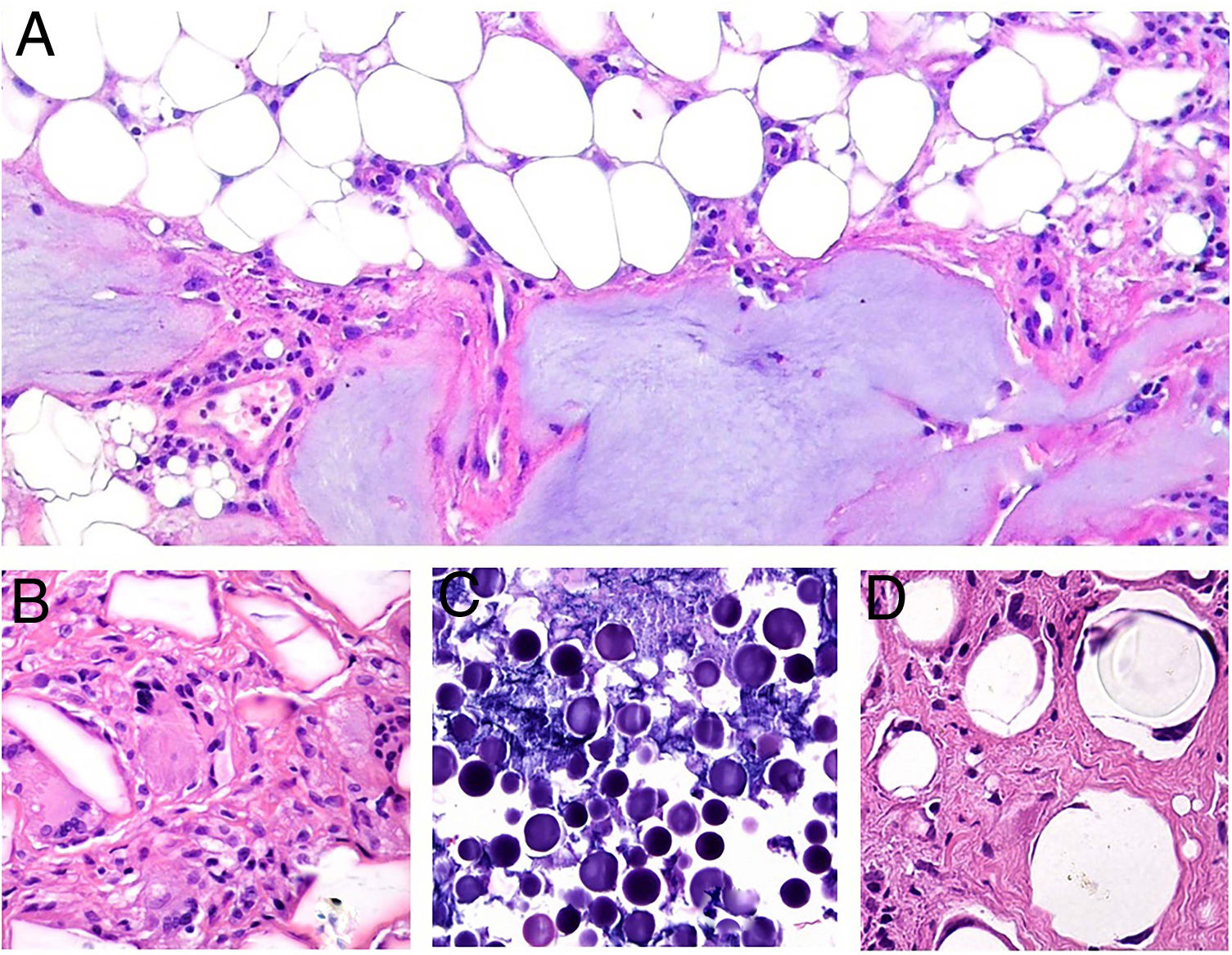
Clinical manifestations. NL typically affects middle-aged adults and is more common in women.15–17 Clinically, it is characterized by violaceous or erythematous papules and nodules, which progress to form coalescent yellowish-brownish plaques, which usually have an atrophic, telangiectatic center. Secondary ulceration is common15,17 (Fig. 10A). NL lesions are mainly found in the pretibial region, but other areas have been described, including the upper limbs, face, and scalp.15,17,18 Although lesions tend to be asymptomatic, they can causing itching, dysesthesia, and even pain.18 They may be resistant to multiple treatments,18,19 and, at least in some cases, patients who achieve improved control of their DM may experience partial or complete remission.16
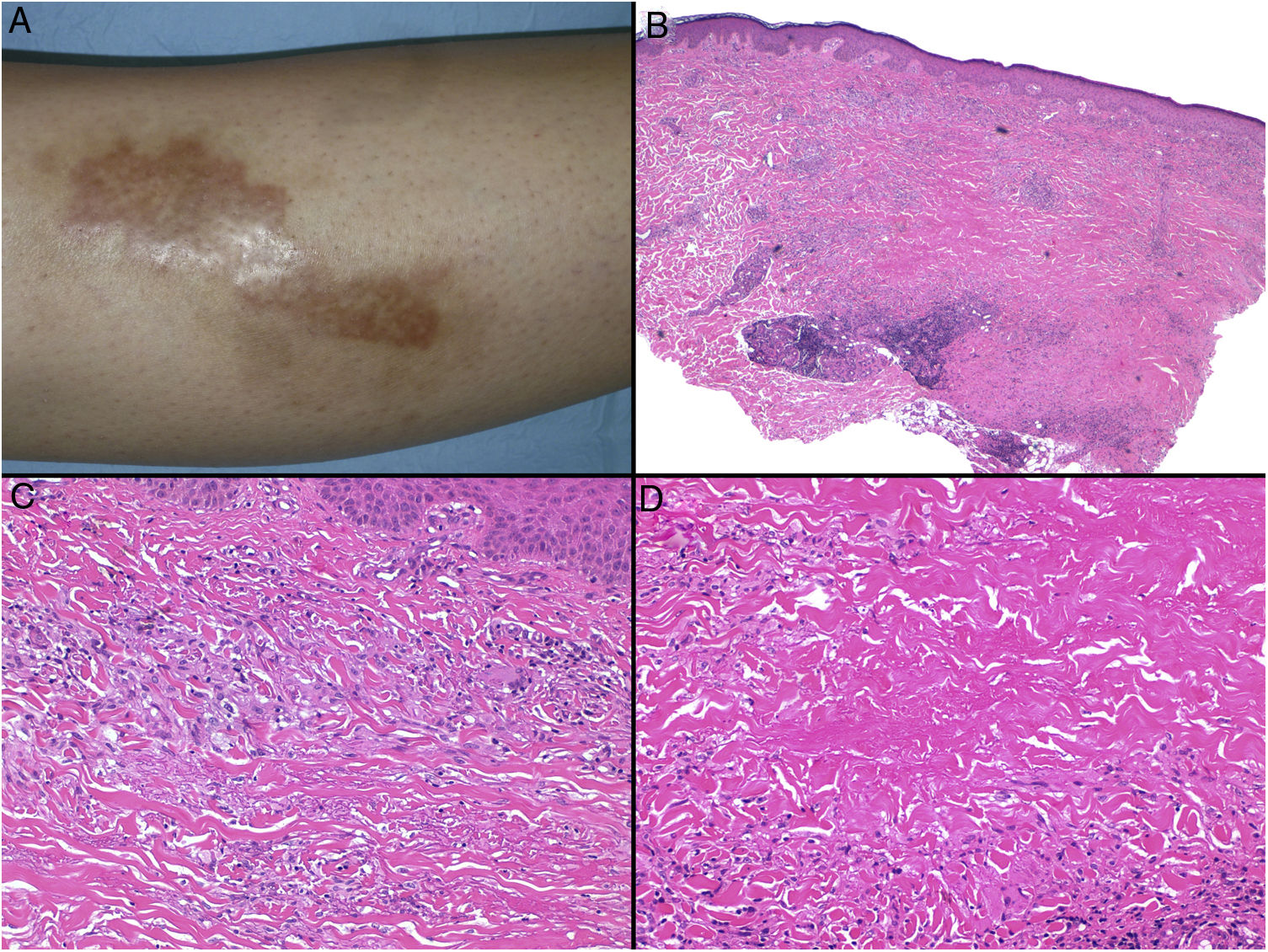
Necrobiosis lipoidica. A, Erythematous-yellowish infiltrative plaque with a wax-like appearance. The center of the plaque is paler and has an atrophic appearance. B, Panoramic view showing a focal inflammatory infiltrate arranged in layers that are more or less parallel to the epidermis, together with areas of thickened, intensely eosinophilic collagen. Note the periadnexal lymphoid aggregates in the deep part of the biopsy specimen (deep dermis) (H&E, original magnification x40). C, Higher-magnification view showing an interstitial granulomatous inflammatory infiltrate arranged between bundles of thickened, degenerated collagen. Several multinucleated histiocytes are also visible (H&E, original magnification x200). D, High-magnification view of the other part of the biopsy specimen showing a palisaded granuloma centered by an area of clearly degenerated collagen (H&E, original magnification x400). H&E indicates hematoxylin-eosin.
Histopathology. Typical histopathologic findings are observed at the active, inflammatory edge of NL lesions. They include an interstitial granulomatous infiltrate centered in the reticular dermis with frequent involvement of the subcutaneous tissue in the form of septal panniculitis (Fig. 10B). This infiltrate is characteristically stacked parallel to the epidermis, alternating with layers of degenerated collagen15,16,20 (Fig. 10C). This pattern is more common in NL associated with DM. Additional findings include areas of palisaded granulomas, with a center composed of necrobiotic collagen and multinucleated giant cells15,19,20 (Fig. 10D). Mucin deposits are absent or scarce. Lymphoid aggregates and a superficial perivascular infiltrate in the adjacent dermis may also be observed; vessels usually have a thickened wall and a slightly narrowed lumen due to the presence of material that stains positively with PAS (mainly in DM-associated NL).15,16,20,21 Adipophilin staining can confirm the presence of extracellular lipid deposits.21 The main entities to consider in the histologic differential diagnosis are other granulomatous disorders and forms of septal panniculitis.
Necrobiotic XanthogranulomaDefinition. Necrobiotic xanthogranuloma (NXG) is a form of non-Langerhans cell histiocytosis, often associated with paraproteinemia. It occurs in the context of plasma cell dyscrasias and other lymphoproliferative disorders.22
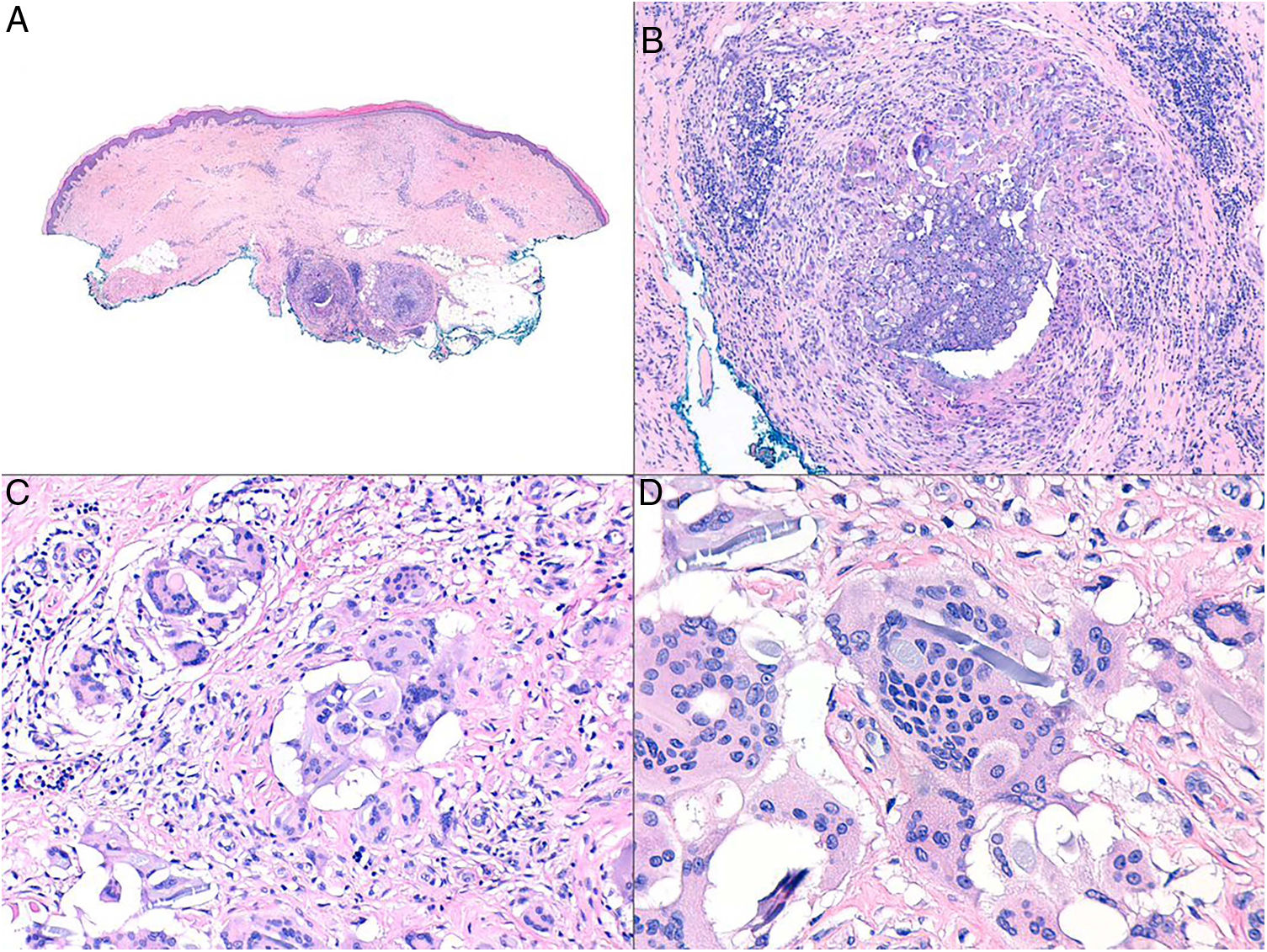
Clinical presentation. NXG presents at a mean age of 61 years and is characterized by often multiple yellow-orange, red, or violaceous lesions in the form of papules, plaques, and less frequently, nodules (Fig. 11).23–25 The most common location is the face, in particular the periorbital region, followed by the limbs and the trunk. Extracutaneous NXG is mainly ocular and has been described in close to 30% of cases.25
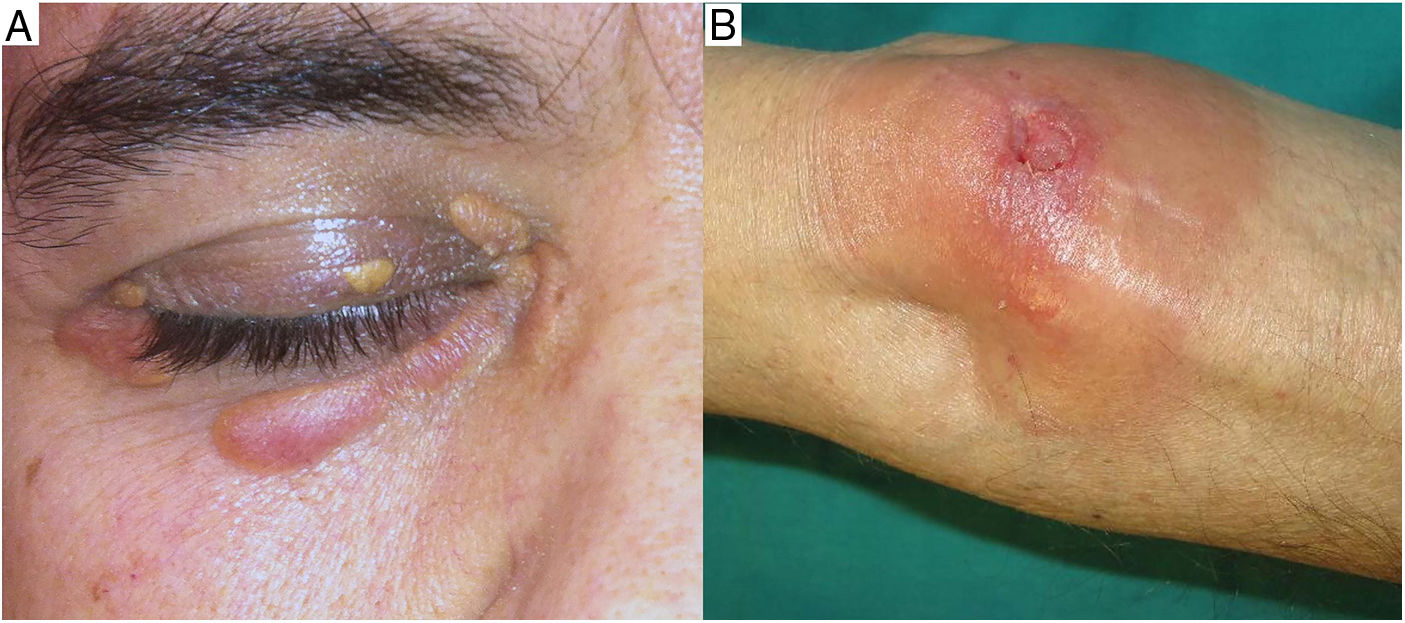
The blood work-up shows lipid alterations, particularly elevations in cholesterol and low-density proteins, low C4 levels, and cryglobulins.24,25
Histopathology. The histopathologic findings in NXG are similar to those seen in NL, with “red” granulomas associated with eosinophilic necrobiosis and a band-like histiocytic infiltrate in the dermis and subcutaneous tissue. The histiocytic infiltrate shows foamy cells and giant cells (Touton and/or foreign body giant cells) associated with nodular lymphocytic aggregates and numerous plasma cells and no evidence of light-chain restriction.26 Extracellular cholesterol crystals are also observed (Fig. 12).27 In one study, immunohistochemical staining showed positive results for extracellular and intracellular adipophilin, contrasting with findings for NL where positive results are only observed for areas of necrobiosis.28
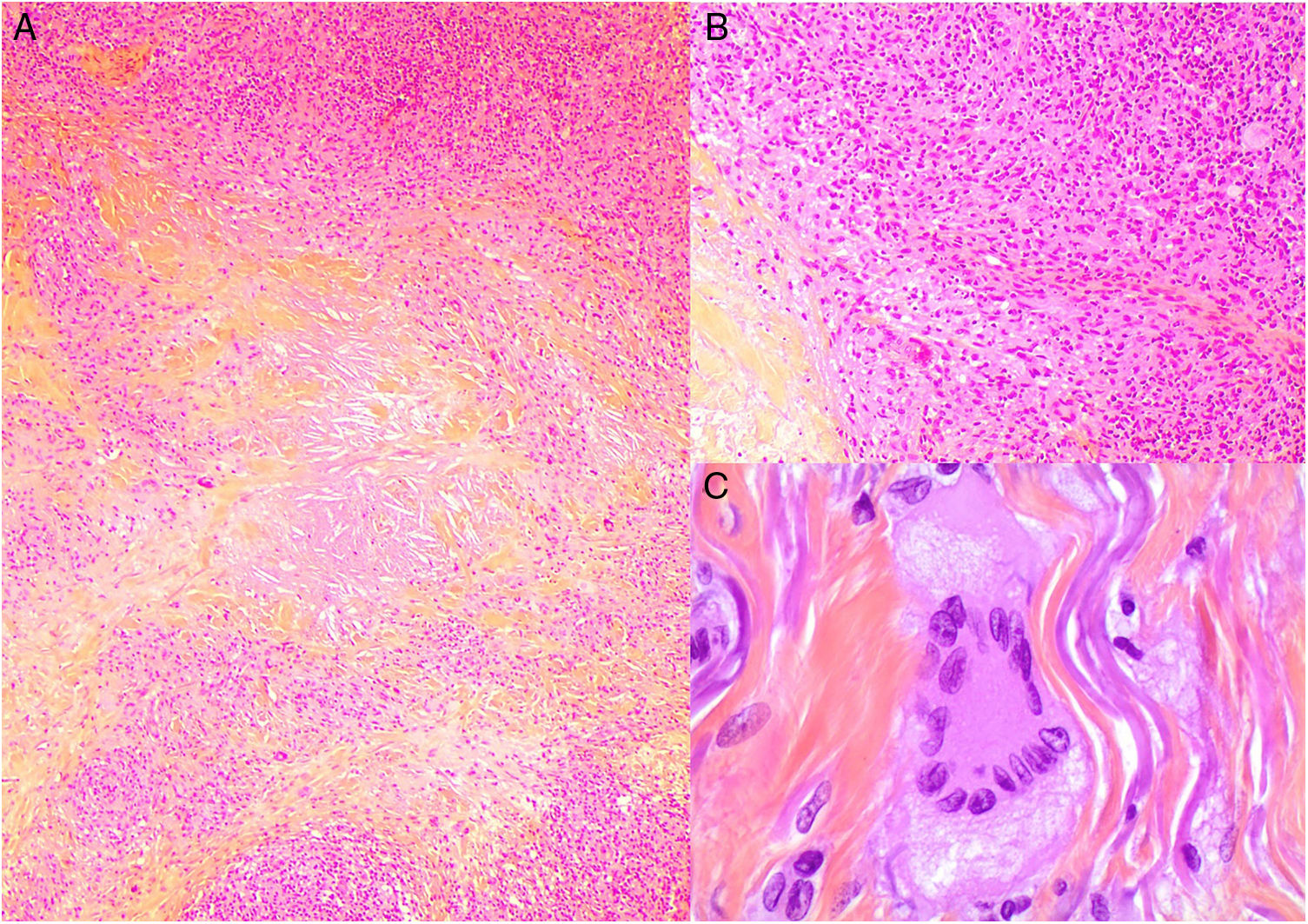
Histologic appearance of xanthogranuloma. A, Granulomatous infiltrate surrounding a central area of eosinophilic necrobiosis with associated cholesterol crystals (H&E, original magnification x40). B, Histiocytic infiltrate accompanied by lymphoid nodules and plasma cells (H&E, original magnification x100). C, Detail of Touton cell (H&E, original magnification x400). H&E indicates hematoxylin-eosin.
According to the recently published diagnostic criteria for NXG25 (Table 1), a diagnosis can be established if both major criteria and at least 1 minor criterion in the absence of foreign body implantation, infection, or any other identifiable cause are met.
Diagnostic Criteria for Necrobiotic Xanthogranuloma Proposed by Nelson et al.25 Both major criteria and 1 minor criterion are needed for diagnosis.
| Major criteria |
| 1. Cutaneous papules, plaques, and/or nodules, most often yellow or orange in color |
| 2. Histopathological features demonstrating palisaded granulomas with lymphoplasmacytic infiltrate and zones of necrobiosis. Characteristic features that are variably present include cholesterol clefts and/or giant cells (Touton or foreign body) |
| Minor criteria |
| 1. Paraproteinemia, most often IgG-κ, plasma-cell dyscrasia, and/or other associated lymphoproliferative disorder |
| 2. Periorbital distribution of lesions |
Definition. Inflammatory bowel disease (IBD) consists of 2 main entities: Crohn disease and ulcerative colitis. Although manifestations are mainly gastrointestinal, up to 40% of patients can experience other manifestations, particularly those with Crohn disease.29 IBD can affect practically all organ systems, including the eyes, skin, lungs, kidneys, liver, and the immune, hematologic, and cardiovascular systems.29 The skin is one of the main organs affected; between 10% and 22% of patients with Crohn disease or ulcerative colitis develop skin manifestations.29,30
Clinical presentation. Mucocutaneous manifestations associated with IBD have been divided into 5 categories according to their pathophysiologic association with the underlying disease:
- 1
Specific manifestations with the same histologic features as the underlying IBD
- 2
Mucocutaneous disorders associated with IBD, the most common being erythema nodosum, pyoderma gangrenosum, and aphthous stomatitis31
- 3
Reactive IBD manifestations due to immunologic mechanisms triggered by antigens shared by intestinal bacteria and the skin
- 4
Mucocutaneous conditions due to IBD treatment
- 5
Manifestations due to nutritional malabsorption
Metastatic Crohn disease involves the development of specific noncaseating skin granulomas at a distance from the gastrointestinal tract.29 The most frequently involved sites are the lower extremities and intertriginous areas, although lesions can occur anywhere (Fig. 13). The clinical manifestations of metastatic Crohn disease consist of erythematous plaques, nodules, abscesses, fistulas, and ulcers. No clear correlation has been established between the severity of Crohn disease and metastasis.
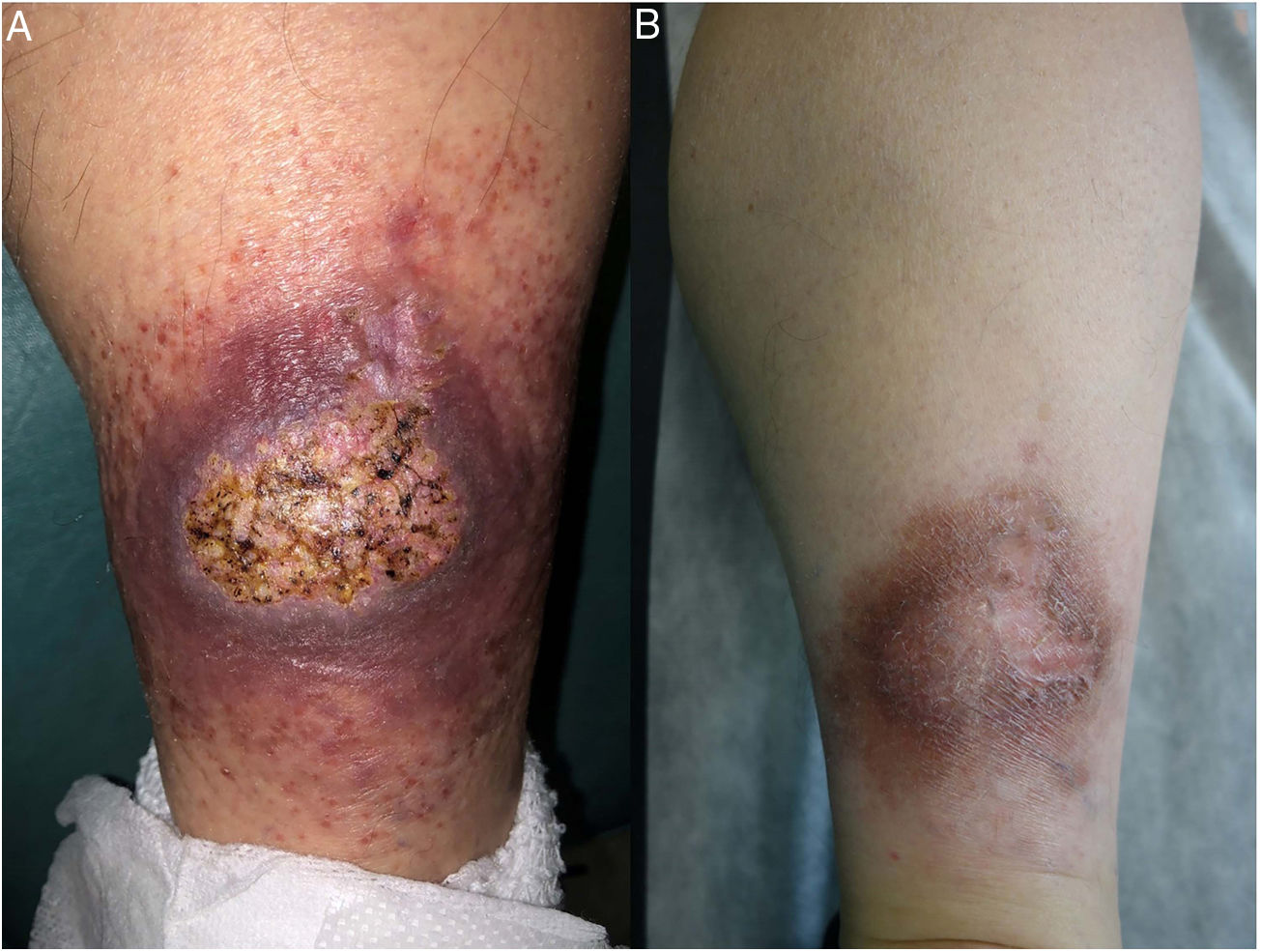
Histopathology. Histopathologic findings in metastatic Crohn disease can resemble those seen in intestinal Crohn disease and include sterile, noncaseating granulomas with foreign body giant cells or Langhans cells, epithelioid histiocytes, and plasma cells (Figs. 14A,B).32 On occasions, granulomas are seen around normal-looking blood vessels. They are often associated with a lymphocytic infiltrate in the reticular dermis, possibly extending into the subcutaneous tissue. Less common findings are collagen degeneration, eosinophilic infiltration, ulceration, dermal edema, and lichenoid dermatitis.
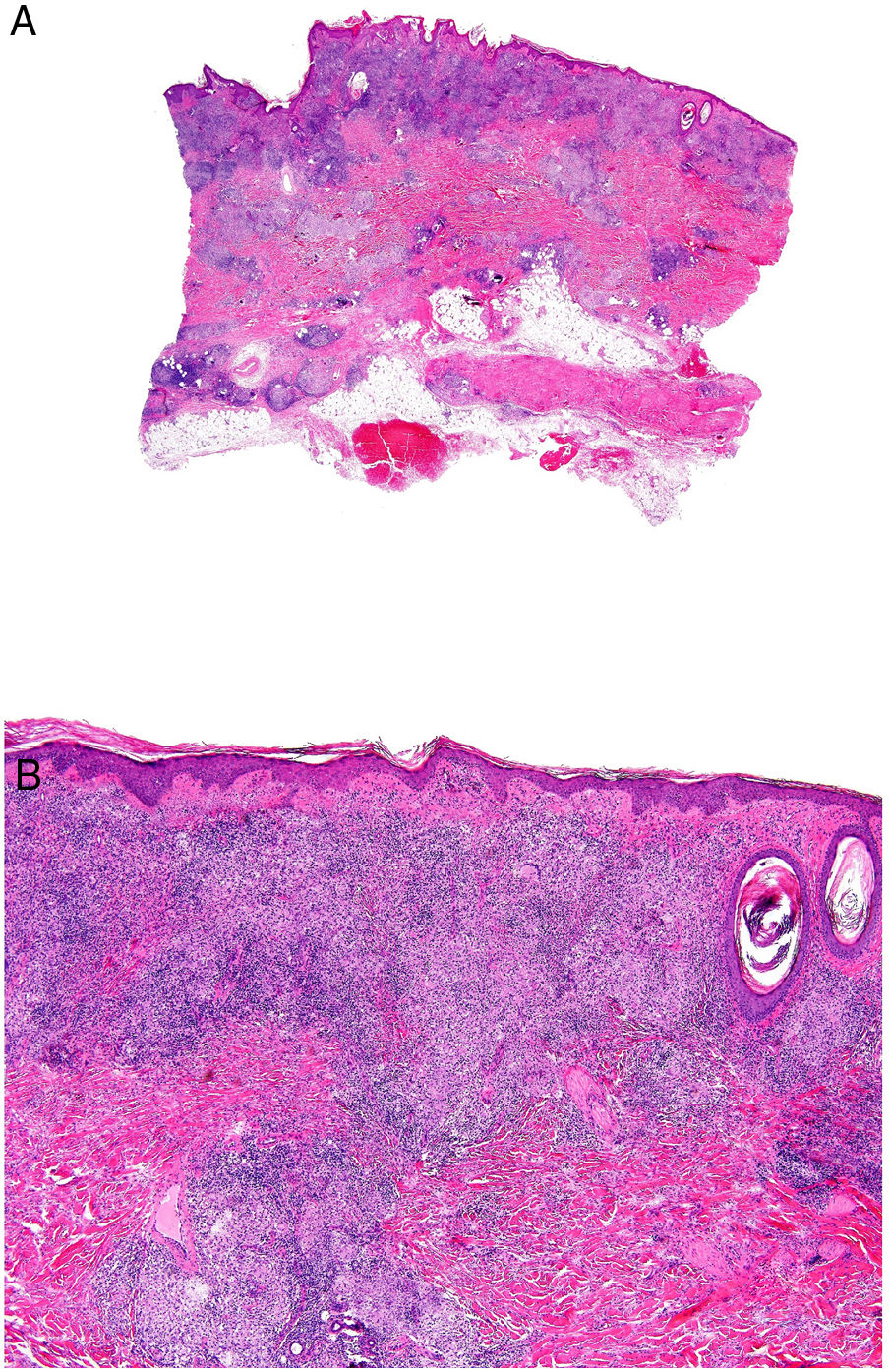
Crohn disease. A, Granulomatous dermatitis with multiple granulomas in the reticular dermis, with sparing of the epidermis and papillary dermis (H&E, original magnification x20). B, The granulomas are similar to those seen in the digestive tract: noncaseating with multinucleated giant cells and possibly a dense lymphocytic infiltrate (H&E, original magnification x40). H&E indicates hematoxylin-eosin.
Definition. Granulomatous slack skin is a very rare subtype of mycosis fungoides with an indolent course (5-year survival of almost 100%). Its diagnosis is based on clinical-pathologic data.33
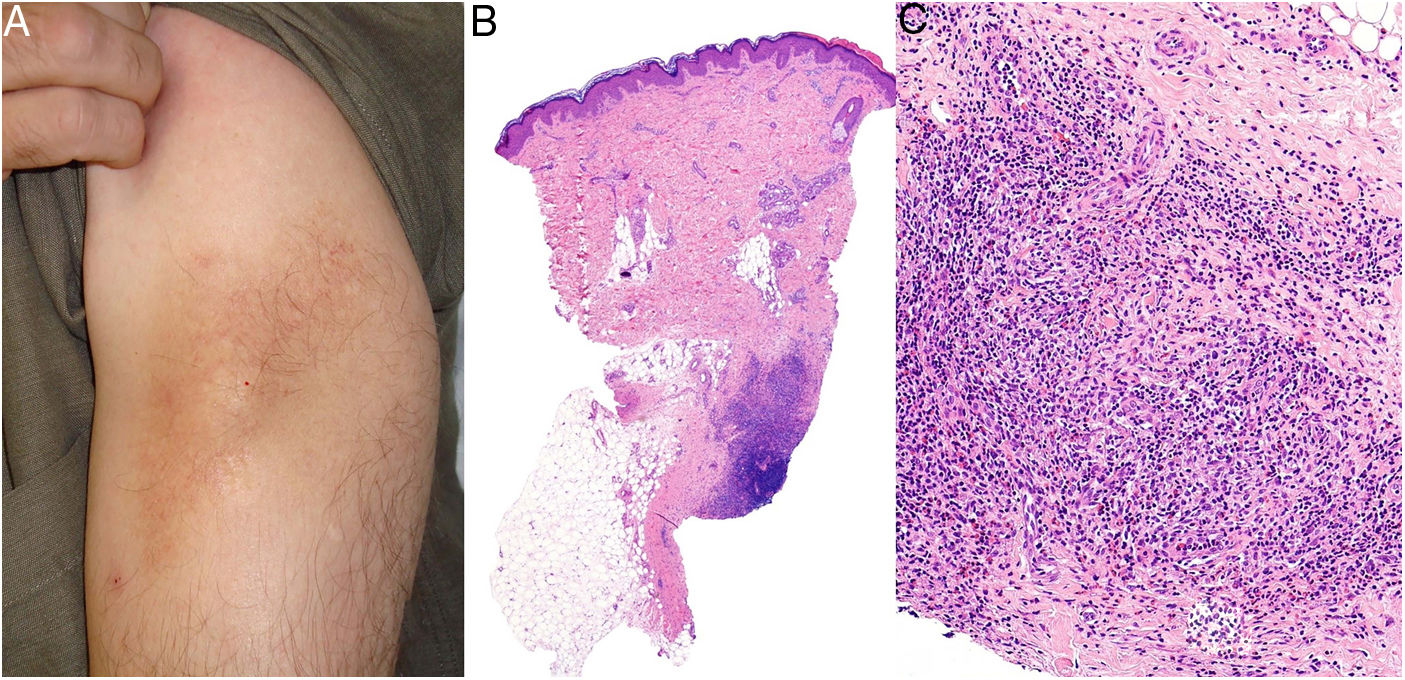
Clinical presentation. More common in young patients, granulomatous slack skin is characterized by erythematous plaques and papules with a flaccid, sagging appearance in flexural areas, with a predilection for the axillae and groin. It grows slowly over years (Fig. 15A) and may occur in association with a systemic hematologic disease, such as Hodgkin lymphoma, acute myeloid leukemia, or Langerhans cell histiocytosis.
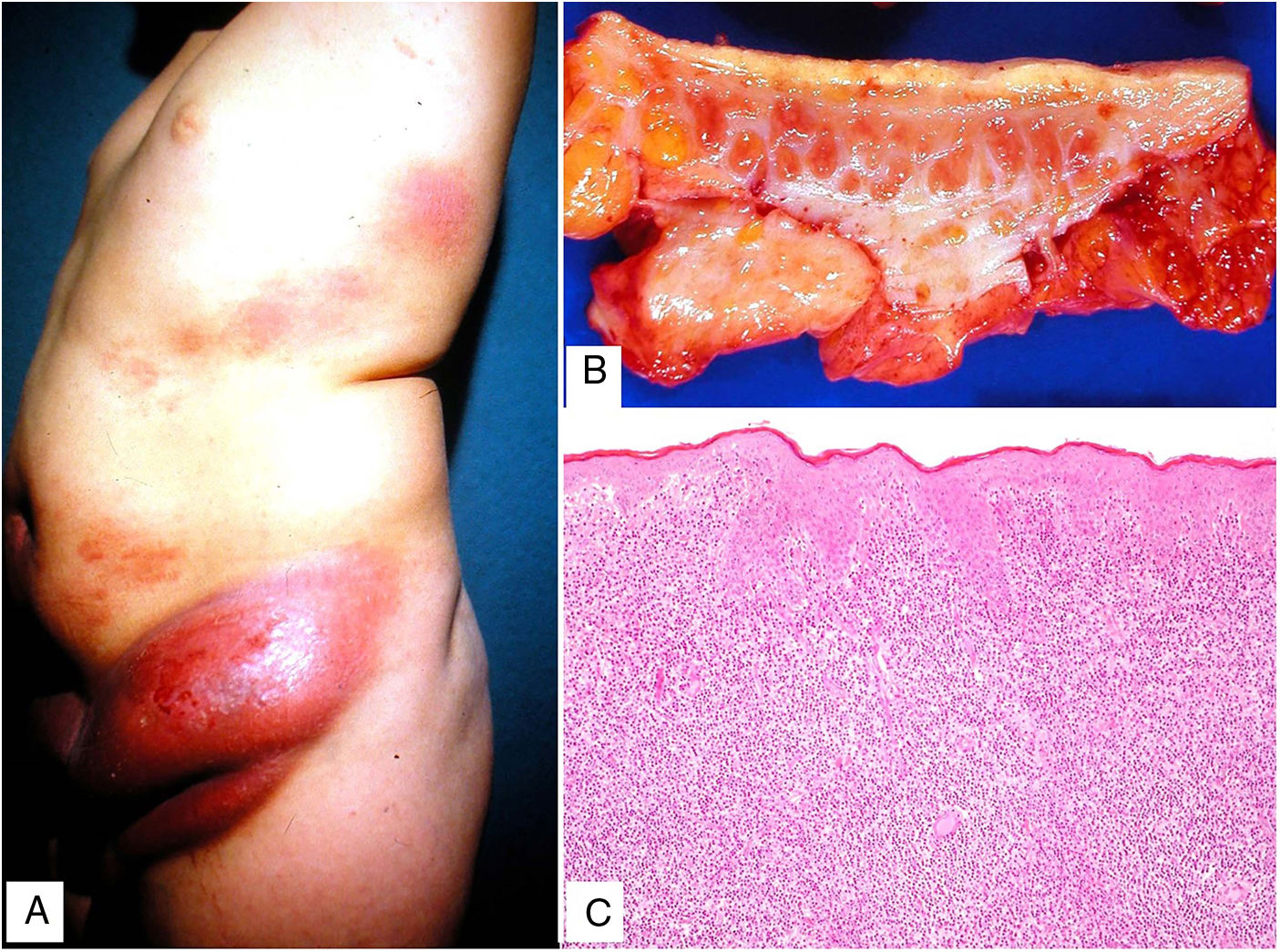
Histopathology. Histopathologic findings include a dermal infiltrate of small atypical lymphocytes (T helper immunophenotype: CD3+, CD4+, and CD8− ) that can extend into the subcutaneous tissue (Fig. 15B,C) and is accompanied by numerous multinucleated giant cells (Fig. 16A,B) showing elastophagocytosis and lymphophagocytosis (Fig. 16C,D). Findings in early-stage disease are similar to those seen in conventional mycosis fungoides. Polymerase chain reaction shows T-cell receptor gene rearrangement.34
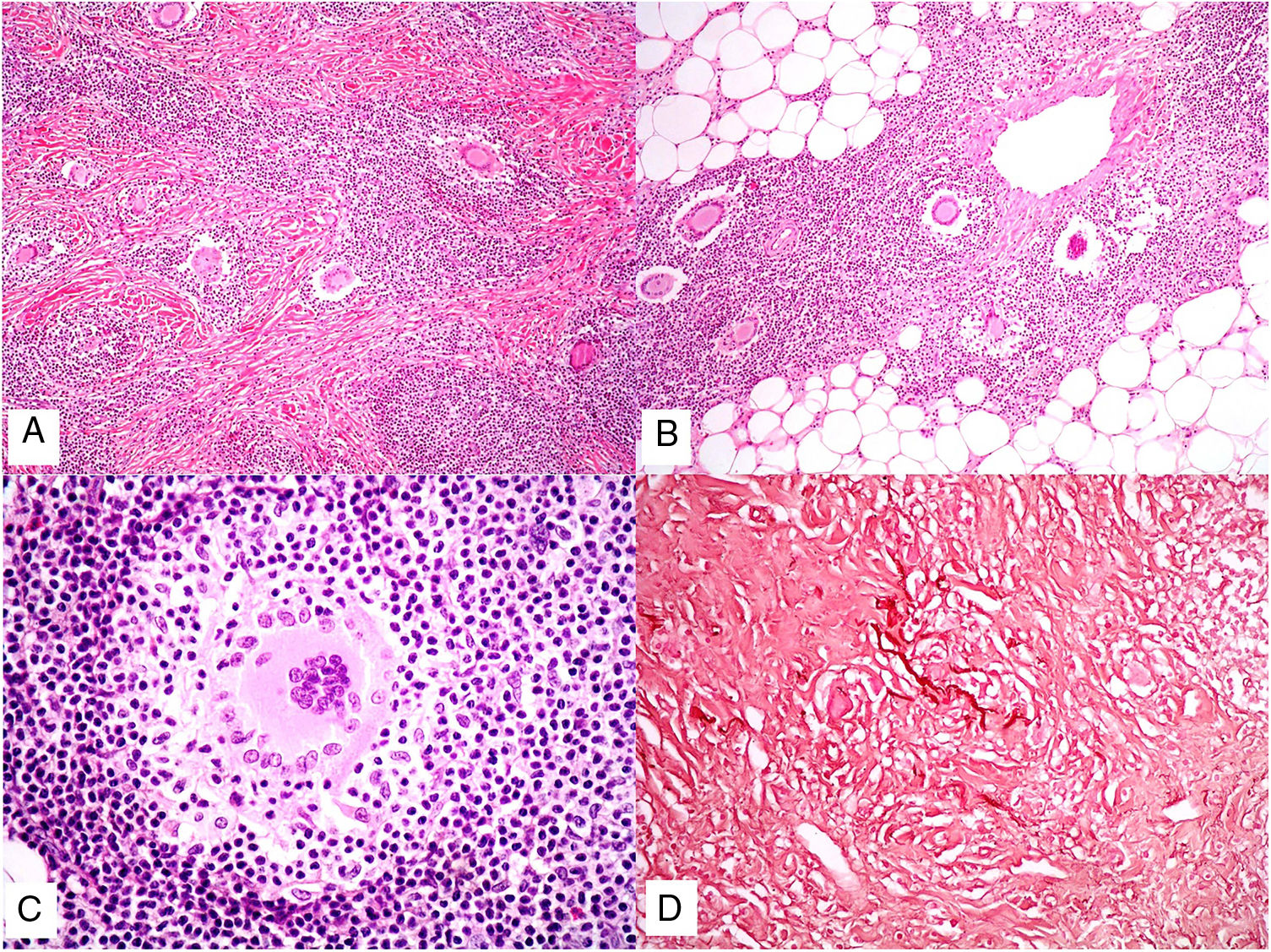
Granulomatous slack skin. A, Lymphoid infiltrate in the dermis with numerous scattered multinucleated giant cells (H&E, original magnification x40). B, Lymphoid infiltrate in the septae of the subcutaneous tissue with multinucleated giant cells (H&E, original magnification x40). C, Detail of a giant cell with lymphophagocytosis (H&E, original magnification x200). D, Fragmentation and loss of elastic fibers (orcein x100). H&E indicates hematoxylin-eosin.
Granulomatous slack skin should not be confused with granulomatous mycosis fungoides (giant cells with fewer nuclei and absence of evident lymphophagocytosis). Focal nucleated histiocytes due to the effects of treatment or the rupture of pilosebaceous follicles are observed in the tumor phase of mycosis fungoides. Integration of clinical and pathologic findings is necessary in all cases.33,35
Granulomatous Mycosis FungoidesDefinition. Granulomatous mycosis fungoides is an exclusively histopathologic variant of mycosis fungoides characterized by the presence of granulomas.36
Clinical presentation. The cases described to date have involved spots, plaques, and tumors with no clinical evidence suggestive of a granulomatous infiltrate (Fig. 17A). Although the presence of a granulomatous infiltrate was initially attributed a protective role in mycosis fungoides, it now appears to be associated with a worse prognosis than nongranulomatous forms.
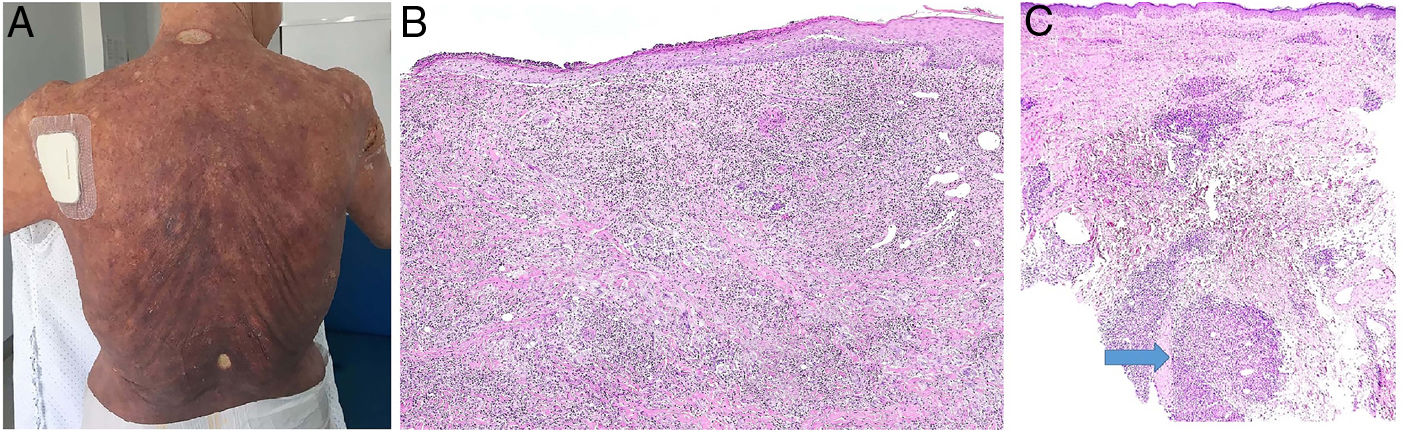
A, Granulomatous mycosis fungoides. Extensive (histopathologically granulomatous) mycosis fungoides on the back of a 77-year-old woman with long-standing disseminated lesions. Note the focally ulcerated tumor plaques and nodules (photo courtesy of Dr César Perez-Vega). B, Granulomatous mycosis fungoides. In addition to the atypical lymphocytes in the epidermis, note the dermal infiltration including giant cells and granulomas of variable texture (H&E, original magnification x20). C, Non-mycosis fungoides cutaneous T-cell lymphoma. Interstitial dermal infiltrate in a patient with peripheral T-cell lymphoma. Note the formation of a deep dermal nodule containing confluent sarcoid-like epithelioid granulomas (arrow) associated with lymphoma cells (H&E, original magnification x10). H&E indicates hematoxylin-eosin.
Histopathology. Considerable variations have been observed in the number and characteristics of the granulomas described to date, with reports ranging from isolated multinucleated giant cells to poorly defined granulomatous infiltrates and compact epithelioid granulomas. The granulomas tend to be widely distributed and occupy the full thickness of the dermis (Fig. 17B). There have been rare reports of an exclusively superficial pattern.37 The combination of subcutaneous tissue involvement and sagging skin is characteristic of granulomatous slack skin, which, as mentioned earlier, is one of the 3 variants of mycosis fungoides recognized in the World Health Organization classification. Integration of clinical and pathologic findings is essential for distinguishing between granulomatous mycoides fungoides and granulomatous slack skin. Cells expressing CD123 have been described in granulomatous mycosis fungoides.38
Other LymphomasDefinition. Granulomatous reactions are uncommon (1.6%–1.8%)39 but well-known occurrences in low- or high-grade primary or secondary cutaneous B-cell and T-cell lymphomas.
Clinical presentation. The clinical presentations described to date are nonspecific and include rashes, erythematous plaques, arciform40 or annular40,41 asymmetric42 plaques, and nodules.43
Histopathology. Granulomatous reactions may be observed in low- or high-grade primary or secondary cutaneous B-cell and T-cell lymphomas. The lymphocytic population is therefore highly variable and is determined by the underlying tumor. In general, primary cutaneous lymphomas show a dermal or dermal-hypodermal reaction of variable intensity depending on the extent of lymphoma invasion44 (Fig. 17C). On occasions, the abundance of granulomas can lead to confusion with granulomatous dermatitis, delaying the diagnosis of lymphoma.39,45 Reactions have been described in CD30+ cutaneous T-cell lymphoma, panniculitis-like T-cell lymphoma, primary marginal zone B-cell lymphoma, and follicle-center lymphoma.39,44
Two types of granulomatous reactions have been described in systemic lymphomas with secondary cutaneous involvement: one showing tumor cells and the other showing no signs of lymphoma involvement. The histologic patterns include tuberculoid-type, palisaded and necrobiotic, and sarcoid-type granulomas.40 It remains unclear whether the granulomas are caused by the presence of the lymphoma or represent a particular immune response. Opportunistic infections must be ruled out. The main systemic lymphomas with secondary cutaneous involvement are peripheral T-cell lymphoma, angioimmunoblastic T-cell lymphoma, diffuse large B-cell lymphoma, Hodgkin lymphoma, adult leukemia /T-cell lymphoma, Sézary syndrome, and histiocyte-rich T-cell lymphoma.39,41,43,44
The prognostic significance of granulomas in primary cutaneous lymphomas or systemic lymphomas with cutaneous involvement is unknown. 45
Alopecia With Granulomatous ReactionsDefinition. Different granulomatous patterns have been described in several types of alopecia. In some cases, the inflammation is directly related to the cause of the alopecia, such as an infection. In other cases, it is one of several findings in a not-yet clear or unknown disease. It may also be a response to the exit of the hair shaft or to other parts of the hair follicle following rupture.
Clinical presentation. Most granulomatous reactions cause the destruction of adjacent structures. In the case of hair follicles, thus, they cause irreversible damage to the hair follicle bulge and hence destroy the follicle, resulting in irreversible scarring alopecia. Other clinical manifestations depend on the type of alopecia.
Central centrifugal cicatricial alopecia (CCCA) presents as centrifugally spreading hair loss of varying intensity on the vertex of the scalp. Neutrophilic cicatricial alopecia (folliculitis decalvans, dissecting cellulitis of the scalp, and acne keloidalis), in turn, presents as edematous, indurated areas of skin that progress to cause further hardening and scarring and irreversible hair loss. The areas affected vary according to the condition involved. The vertex and occipital scalp are affected in folliculitis decalvans, the vertex and crown in dissecting cellulitis of the scalp, and the posterior neck in acne keloidalis nuchae.
Histopathology. Asymmetric thinning of the upper portion of the hair follicle has an essential role in CCCA, as it causes rupture of the follicle wall, leading to penetration of the hair shaft into the dermis and triggering a granulomatous foreign body reaction (Fig. 18A).
![A, Central centrifugal cicatricial alopecia. Foreign body granuloma formed in response to hair shaft fragments and keratin lamellae (H&E, original magnification x100). B and C, Folliculitis decalvans. Foreign body granulomatous reactions to hair shaft fragments may be observed in advanced stages (H&E, original magnification x20 [B] and x100 [C]). H&E indicates hematoxylin-eosin.](https://static.elsevier.es/multimedia/15782190/0000011200000008/v2_202110201006/S1578219021002110/v2_202110201006/en/main.assets/gr18.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AsJ77o2ZFetlwOuU3sEneYXoKNPw/cBP1mRtgus4N5LwD6q9E4jbdt0340o41hiphkRsbOPV+lDVbOu9X+rnjqHf6Vmc8YbsTKmTPn786NmzTK8LWxV9Ur8ByFAc27Pjf/P6ZOaeFno6VtyhaD+wlDXNmfgcIUyt5KKP7V7xR+rBWlNMR4mHOIgvVetapBr07m4OR/Aeny+XaOLxyfMiuGoILppB8sWNn89GpnVM/y1p)
A, Central centrifugal cicatricial alopecia. Foreign body granuloma formed in response to hair shaft fragments and keratin lamellae (H&E, original magnification x100). B and C, Folliculitis decalvans. Foreign body granulomatous reactions to hair shaft fragments may be observed in advanced stages (H&E, original magnification x20 [B] and x100 [C]). H&E indicates hematoxylin-eosin.
Granulomatous inflammatory infiltrates may be observed in advanced neutrophilic cicatricial alopecias (folliculitis decalvans, dissecting cellulitis of the scalp, and acne keloidalis) (Fig. 18B). The granulomas are foreign body granulomas, usually triggered by hair shaft fragments (Fig. 18C). Abscesses can sometimes form, but will show negative results for fungi and mycobacteria.
Antineutrophilic Cytoplasmic Antibody–Associated VasculitisDefinition. Antineutrophilic cytoplasmic antibody–associated vasculitis comprises a heterogeneous group of rare autoimmune diseases that cause inflammation of the blood vessels and have a range of clinical manifestations. There are 3 main entities: granulomatosis with polyangiitis (GPA), formerly known as Wegener granulomatosis; eosinophilic granulomatosis with polyangiitis (EGP), formerly known as Churg-Strauss syndrome; and microscopic polyangiitis. Other ANCA-associated diseases are drug-induced vasculitis and renal-limited vasculitis.46
Clinical Presentation- 1
GPA usually occurs in the fourth and fifth decades of life and is characterized by upper respiratory tract symptoms, such as persistent rhinorrhea, sinus pain, cough, and hemoptysis. Skin manifestations occur in 30% to 50% of cases and can sometimes be the presenting symptom. They consist of symmetrically distributed papulonecrotic lesions on the elbows, knees, and occasionally, buttocks.47
- 2
EGP affects middle-aged adults and is more common in men. According to the American College of Rheumatology classification, a patient must have 4 of the following 6 findings to be diagnosed with EGP: asthma, eosinophilia greater than 10%, paranasal sinusitis, pulmonary infiltration, histologically proven vasculitis, and mononeuritis multiplex.48 Cutaneous manifestations include hemorrhagic lesions (ranging from petechiae to palpable purpura and extensive ecchymosis, sometimes accompanied by necrotic ulcers), and cutaneous or subcutaneous nodules.
- 3
Microscopic polyangiitis is a systemic small-vessel vasculitis typically associated with focal necrotizing crescentic glomerulonephritis and antineutrophil cytoplasmic antibodies against serum myeloperoxidase (p-ANCA). It mainly affects men aged over 50 years. The most common clinical manifestation is nephropathy, which presents with microhematuria, proteinuria, or oliguric acute kidney failure. It is accompanied by prodromic symptoms, such as fever, myalgia, arthralgia, and sore throat. At least 30% to 40% of patients with microscopic polyangiitis develop skin manifestations, such as erythematous macules, reticularis livedo, palpable purpura, splinter hemorrhages, and ulcers.46
- 1
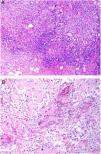
Most biopsies of GPA show nonspecific findings, including perivascular lymphocytic infiltrates. Nonetheless, leukocytoclastic vasculitis and necrotizing granulomatous inflammation are observed in between 25% and 50% of patients.49 The granulomatous infiltrates contain histiocytes, lymphocytes, and giant cells with suppurative necrosis, forming “blue” granulomas with a palisading pattern; they surround necrobiotic collagen and necrotic basophilic detritus intermixed with neutrophils50 (Fig. 19A). None of the findings are pathognomonic of GPA and may be accompanied by true granulomatous vasculitis, acneiform perifollicular granulomatous inflammation, erythema nodosum–like findings, and palisaded neutrophilic dermatosis.47
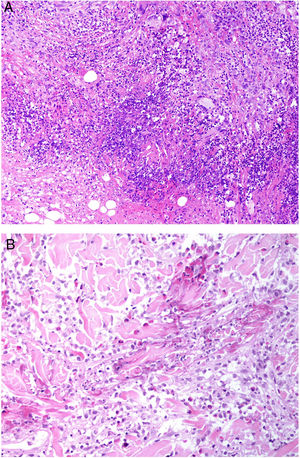 Figure 19.
Figure 19.A, Granulomatosis with polyangiitis. Granulomatous infiltrate with poorly formed granulomas consisting of histiocytes, lymphocytes, and giant cells with suppurative necrosis and traces of necrobiotic collagen (H&E, original magnification x100). B, Eosinophilic granulomatosis with polyangiitis. Granuloma consisting of radially arranged histiocytes around fibers of degenerated collagen with traces of eosinophils (H&E, original magnification x200). H&E indicates hematoxylin-eosin.
- 2
In EGP, areas of skin hemorrhage usually show leukocytoclastic vasculitis with numerous eosinophils.51 In some cases, the dermis shows a characteristic granulomatous reaction characterized by histiocytes and multinucleated giant cells arranged around degenerated collagen fibers with traces of eosinophils (red granulomas)52 (Fig. 19B). These granulomas were initially thought to be characteristic of EGP and were hence called Churg-Strauss granulomas. They are not, however, always present and are not required for a diagnosis. Other conditions in which they are seen are rheumatoid arthritis, lupus erythematosus, lymphoproliferative diseases, subacute bacterial endocarditis, active chronic hepatitis, and IBD. Extravascular granulomas can be found in subcutaneous tissue and may grow to form nodules.
- 3
Leukocytoclastic vasculitis involving arterioles, venules, and capillaries is observed in microscopic polyangiitis. On occasions, necrotizing vasculitis of medium-sized arteries, a characteristic finding in classic polyarteritis nodosa, is seen. A granulomatous inflammatory component is not common in cutaneous lesions.
Definition. Rheumatic fever is a rare childhood disease caused by an abnormal immune response following pharyngeal group A beta-hemolytic streptococcal infection. Its incidence in the western world has decreased dramatically in the past 50 years thanks to the appropriate use of antibiotics.
Clinical presentation. The clinical presentation of rheumatic fever is fever, arthritis, or carditis after streptococcal pharyngitis. A small percentage of patients develop skin manifestations in the form of erythema marginatum and subcutaneous nodules.53 These nodules are asymptomatic, occur on bony prominences, and are often associated with cardiac involvement.
Histopathology. Histopathologic findings include subcutaneous deposits of fibrin and a variable infiltrate of lymphocytes, neutrophils, and plasma cells frequently surrounded by diffuse palisading of histiocytes. These findings are not pathognomonic of rheumatic fever and need to correlated with clinical findings. The main lesions in the differential diagnosis are rheumatoid nodules, which show well-developed palisading of histiocytes and, unlike rheumatic subcutaneous nodules, a central area of dense necrobiosis.
Rheumatoid NodulesDefinition. Rheumatoid nodules are inflammatory cutaneous or subcutaneous lesions that have traditionally been linked to rheumatoid arthritis.
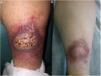
Clinical presentation. The nodules affect up to 30% of patients with rheumatoid arthritis and are one of the main extra-articular manifestations of this disease.54,55 They are most often located near the joints and at pressure points or sites of microtrauma. The main sites affected are the hands, feet, extensor surfaces of the arms and legs (elbows and knees), scalp, ears, buttocks, and back.54,55
Rheumatoid nodules present as firm, noninflammatory nodules that may be attached to the periosteum and deep layers. They range in size from 1 to 5 cm. In the absence of trauma, they are generally asymptomatic and nonulcerated. The number of nodules present varies greatly, although most patients have few nodules or just one (Fig. 20A).
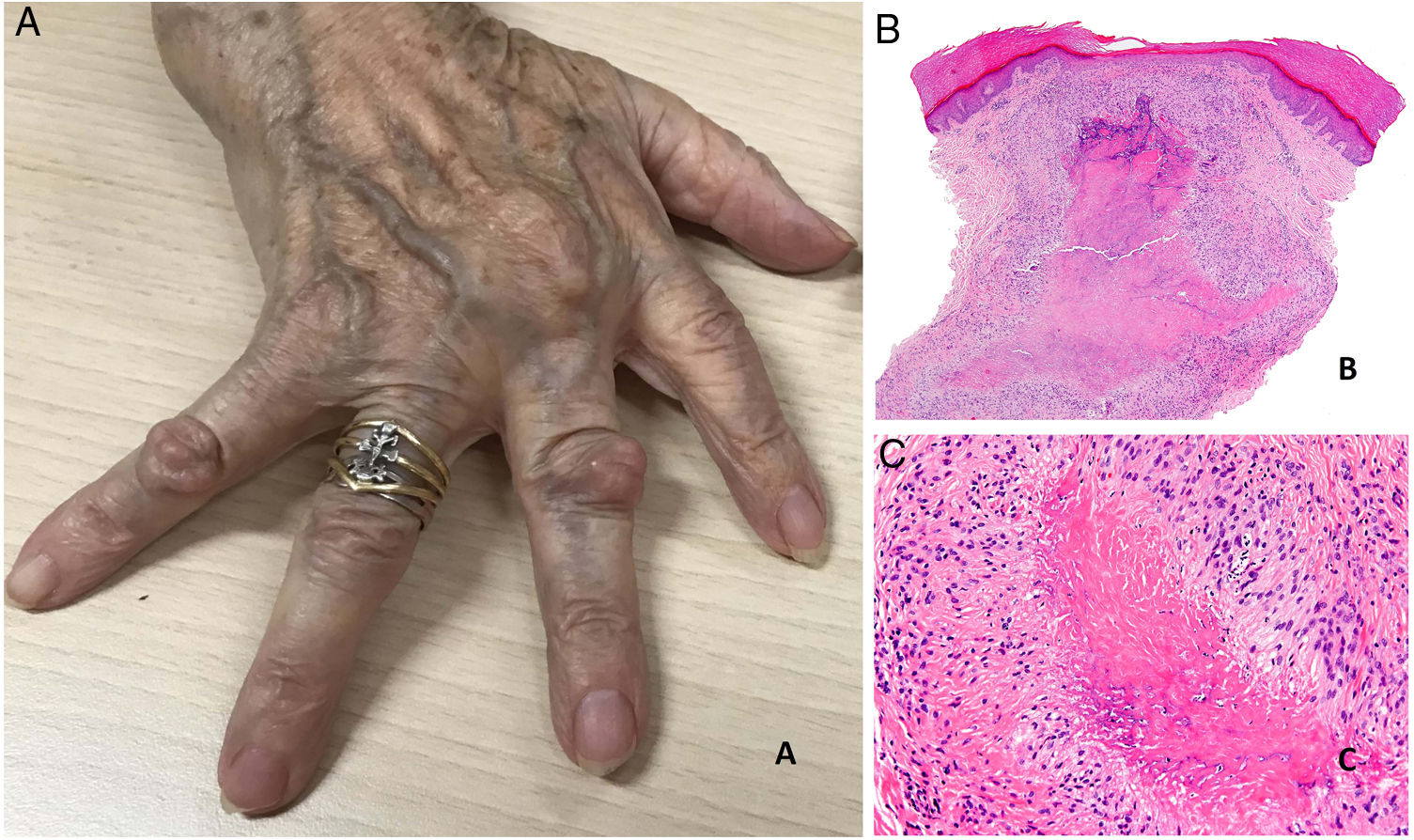
A, Rheumatoid arthritis. Clinical appearance of 2 rheumatoid nodules near the interphalangeal joints of the hands. Note the smooth, whitish surface without signs of inflammation. B, Rheumatoid arthritis. Panoramic histologic view of a rheumatoid nodule, which has reached superficial areas of the dermis and extended into the deep tissues. Note the palisaded granuloma with an intensely eosinophilic necrotic center (H&E, original magnification x20). C, Rheumatoid arthritis. Histologic detail of palisaded granuloma forming the base of the rheumatoid nodule. Note the layer of palisading histiocytes around an intensely eosinophilic center of fibrinoid necrosis (H&E, original magnification x200). H&E indicates hematoxylin-eosin.
Rheumatoid nodules tend to be associated with severe joint disease and high rheumatoid factor titers.55 There have, however, been reports of a more benign clinical form in patients with multiple nodules involving the fingers. This entity has been called rheumatoid nodulosis and is characterized by minimal joint involvement. Treatment with methotrexate can also lead to accelerated rheumatoid nodulosis, characterized by the development of multiple nodules.54,56
As occurs with other extra-articular manifestations of rheumatoid arthritis, rheumatoid nodules can appear at any stage of disease, including early stages.55,56
Exceptionally, rheumatoid nodules can affect internal organs, especially the lungs.56
Histopathology. Rheumatoid nodules have a highly characteristic histologic appearance and represent a granulomatous immune response. They have a central area of necrosis that is strongly eosinophilic due to fibrin deposition, and are surrounded by a well-defined layer of palisading histiocytes frequently rimmed by granulation tissue.54 Because these nodules tend to expand, a confluent multinodular appearance may be observed (Figs. 20 B and C). Rheumatoid nodules are easily distinguishable from granuloma annulare and other palisaded granulomas in the skin, as they are larger, deeper and have a red necrotic center. Other histologic findings include peripheral reactive fibrosis, dystrophic calcifications, and mucin deposits.57 The cytokine profile of rheumatoid nodules is very similar to that observed in the synovial membrane of joints in patients with rheumatoid arthritis; this similarity supports an autoimmune origin, although the precise pathogenic mechanisms remain unclear.56
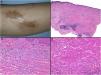
Acne AgminataDefinition. Also known as lupus miliaris disseminatus faciei, acne agminata is a granulomatous inflammatory skin disorder whose origin has been debated for years. It has classically been linked to rosacea, tuberculosis, and sarcoidosis.58
Clinical presentation. Acne agminata usually affects young patients in the form of monomorphic erythematous, brownish, or yellowish papules located in the center and on the sides of the face (Fig. 21). Eyelid involvement, and lower eyelid involvement in particular, is very common. Exceptionally, acne agminata can affect the armpits and upper limbs. The eruption tends to regress leaving residual scarring. Response to tetracyclines, isotretinoin, dapsone, clofazimine, and isoniazid is irregular.59
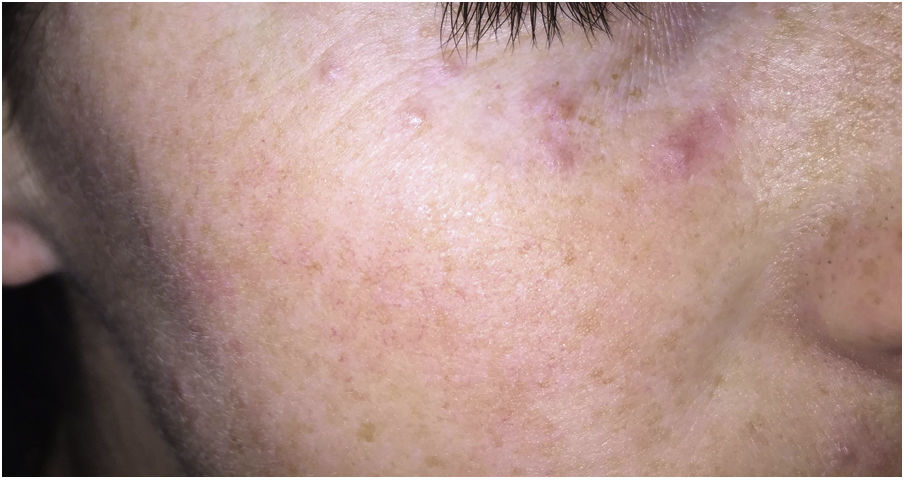
Histopathology. On histology, acne agminata shows a necrotizing epithelioid granulomatous reaction with central caseating necrosis and multinucleated giant cells (Fig. 22). This necrosis may be observed in association with a destroyed hair follicle. In such cases, specific staining may show a ring of elastic fibers in the center of the necrotic focus, possibly corresponding to the hair follicle isthmus. Unlike in rosacea, no associations have been established with Demodex folliculorum.58 Focal vasculitis has been reported in isolated cases.
Rosacea![Acne agminata. Note the necrotic, caseating epithelioid granulomatous response in the dermis (hematoxylin-eosin, original magnification x20 [A], x40 [B], x100 [C], x200 [D]).](https://static.elsevier.es/multimedia/15782190/0000011200000008/v2_202110201006/S1578219021002110/v2_202110201006/en/main.assets/gr22.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6AsJ77o2ZFetlwOuU3sEneYXoKNPw/cBP1mRtgus4N5LwD6q9E4jbdt0340o41hiphkRsbOPV+lDVbOu9X+rnjqHf6Vmc8YbsTKmTPn786NmzTK8LWxV9Ur8ByFAc27Pjf/P6ZOaeFno6VtyhaD+wlDXNmfgcIUyt5KKP7V7xR+rBWlNMR4mHOIgvVetapBr07m4OR/Aeny+XaOLxyfMiuGoILppB8sWNn89GpnVM/y1p)
Definition. Rosacea is a chronic inflammatory disease of the pilosebaceous unit that usually affects middle-aged and older patients and presents as an acneiform papulopustular eruption. It is characterized by increased capillary reactivity that causes persistent erythema and dilated blood vessels. Patients with more advanced stages of disease may develop sebaceous hyperplasia, which can be particularly pronounced on the nose, causing rhinophyma. This condition is more common in men. Although the etiology of rosacea is unknown, the underlying disorder appears to be a reduction in vascular tone with a tendency towards excessive vasodilation triggered by factors such as abrupt changes from cold to heat, exposure to the sun and UV-A radiation, alcohol, very hot or spicy foods, and stressors. Other possible causes include immune system activation and infestation by Demodex mites.60,61
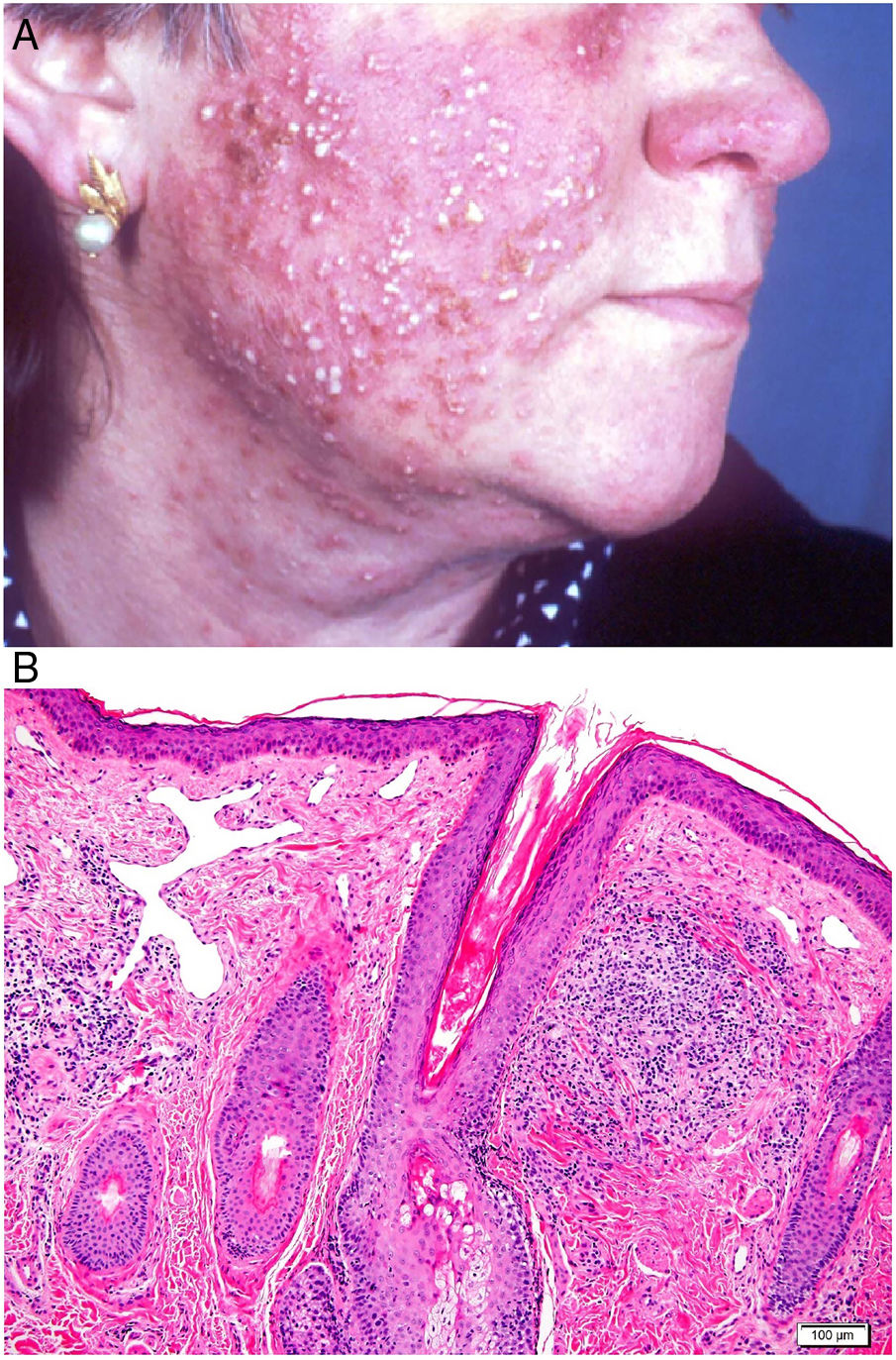
Clinical presentation. Rosacea tends to occur in inflammatory flare-ups with swelling of the skin. Spontaneous remission is very uncommon. Four clinical forms have been described: telangiectatic, papulopustular, granulomatous, and phymatous.62,63 The presenting clinical signs are redness and dilated blood vessels involving the nose, cheeks, and forehead; these are initially intermittent, but they become persistent when telangiectasia develops. In more advanced stages of disease, patients develop symmetric papules and pustules (characteristically without comedones) that cause itching, burning, and stinging (Fig. 23A). Rosacea can occasionally affect the eyes, causing blepharitis, conjunctivitis, episcleritis, iritis, or keratitis.64,65 The granulomatous variant presents with small, hard brown papules. Rhinophyma affects men aged over 40 years old and is characterized by lobulated masses on the tip and the sides of the nose with evident dilation of the pilosebaceous follicles, which are blocked by large amounts of sebum and keratin.
The clinical differential diagnosis should include skin diseases with facial erythema, such as seborrheic dermatitis, lupus erythematosus, dermatomyositis, carcinoid syndrome, vera polycythemia, and acneiform diseases such as acne vulgaris, perioral dermatitis, Gram-negative folliculitis, and steroid acne.
Histopathology. The only histopathologic findings in telangiectatic forms of rosacea are dilation of the dermal lymphatic vessels and a superficial lymphocytic and plasmocytic perivascular infiltrate. The infiltrate in papulopustular forms is denser, involves the deeper layers, and is accompanied by abundant neutrophils around vascular and pilosebaceous structures. Additional findings may include acute folliculitis (in active pustular lesions), follicular plugs, and Demodex mites (20%–50% of cases). The granulomatous variant is characterized by perifollicular granulomas formed by epithelioid cells with a crown of lymphocytes (Fig. 23B) and central necrosis in up to 11% of cases. Rhinophyma is also characterized by sebaceous gland hyperplasia.66
The differential diagnosis should include orofacial granulomatous diseases, such as perioral dermatitis, granulomatous cheilitis, Crohn disease, sarcoidosis, lupus vulgaris, and chronic granulomatous disease.66 Perioral dermatitis and granulomatous cheilitis are microscopically indistinguishable from rosacea and some authors even consider them to be related. Crohn disease granulomas are deeper and affect the vessel walls. Sarcoidosis granulomas are naked, that is, they do not have a crown of lymphocytes. Lupus vulgaris can be diagnosed by the detection of acid-alcohol-fast bacilli, while chronic granulomatous disease occurs in immunodeficient pediatric patients.
Granulomatous Cheilitis and Melkersson-Rosenthal SyndromeDefinition. Granulomatous cheilitis is a rare, immune-mediated non-necrotizing granulomatous disease of the lips. It is considered to be a monosymptomatic or incomplete form of Melkersson-Rosenthal syndrome.
Melkersson-Rosenthal syndrome is also rare and is characterized by a triad of recurrent orofacial edema, facial paralysis, and fissured tongue.
The umbrella term idiopathic orofacial granulomatosis (OFG) is used to describe granulomatous cheilitis and Melkersson-Rosenthal syndrome, which have identical histologic features.
Sarcoidosis, and Crohn disease in particular, may show similar clinical and histologic characteristics to OFG. Nonetheless, strict application of the definition of OFG should be sufficient to rule out other systemic granulomatous diseases.67
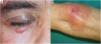
Clinical presentation. The most common clinical presentation of granulomatous cheilitis is recurrent acute, asymptomatic swelling of the lips (Fig. 24A). With time, this swelling becomes firm and indurated and other parts of the face may also be affected. Melkersson-Rosenthal syndrome often presents with incomplete symptoms, particularly in its presenting form. Diagnosis may require long-term follow-up. It has been proposed that orofacial edema must be present for Melkersson-Rosenthal syndrome to be diagnosed.68
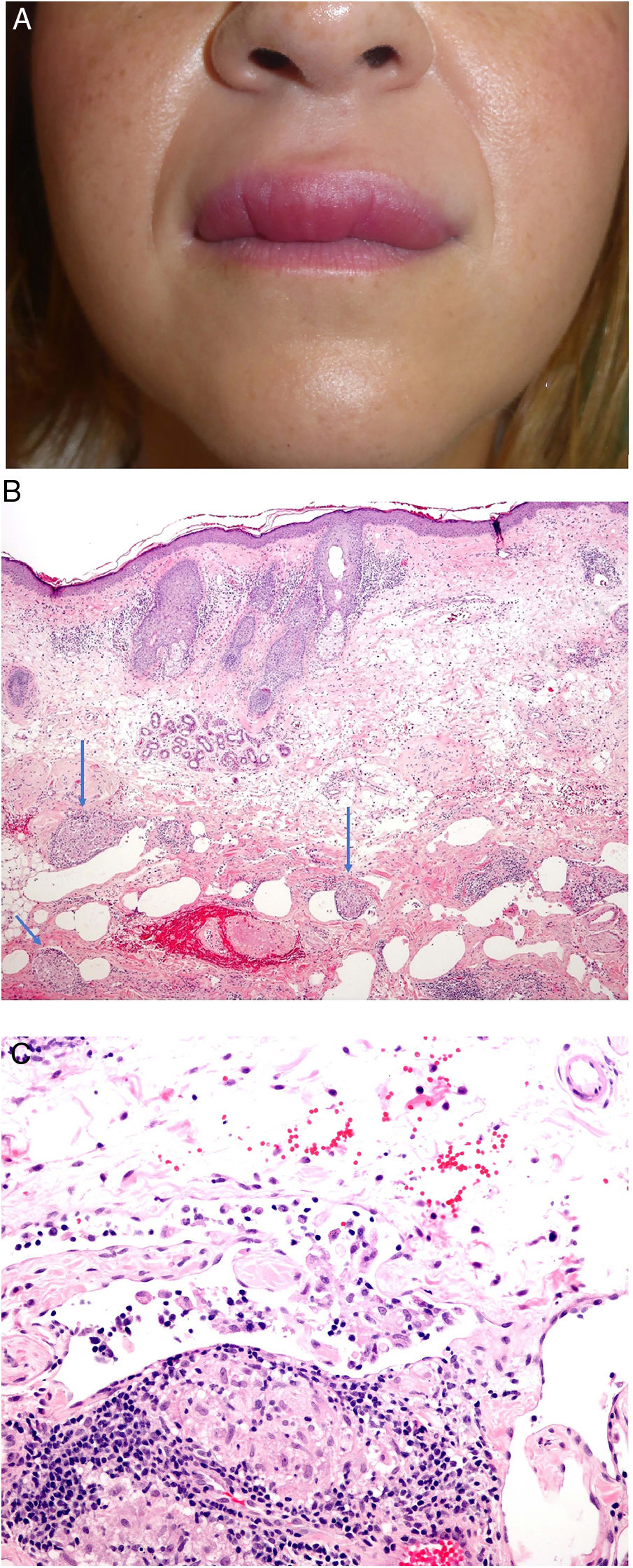
A, Granulomatous cheilitis. Marked swelling of the upper lip. B, Granulomatous cheilitis. Marked swelling of the dermis of the lip, vascular ectasia, mild lymphocytic infiltration, and perivascular and intravascular granulomas (arrows) (H&E, original magnification x40). C, Granulomatous cheilitis. Dilated lymph vessel with intravascular histiocytic aggregates and a perivascular granuloma (H&E, original magnification x200). H&E indicates hematoxylin-eosin.
Histopathology. Reactive changes including hyperkeratosis, acanthosis, and spongiosis with lymphocytic exocytosis are observed in the epithelium of the labial mucosa or epidermis. The chorion and dermis show edema with ectatic blood and lymph vessels and perivascular and interstitial lymphocytic or lymphoplasmacytic and histiocytic infiltrates. Eosinophils may occasionally be present. The most characteristic finding is the presence of non-necrotizing epithelioid granulomas, which are observed in 43% to 82% of cases. Superficial biopsies may not show granulomatous inflammation, as the granulomas are sometimes located in the deep part of the specimen.
Granulomas typically have a loose, noncompact appearance due to the presence of spaces between epithelioid cells. They tend to be small, few in number, disperse, nonconfluent, and naked (not crowned by lymphocytes).69,70 They are often located adjacent to blood and lymph vessels (Fig. 24B). Histiocytic aggregates or granulomas within lymph vessels are occasionally observed (Fig. 24C). This perivascular or intravascular granulomatous inflammation is possibly responsible for blockage of the lymph vessels, which results in lymph stasis, vascular dilation, and ultimately edema.71,72 Small sarcoid granulomas may occasionally be seen. It is important to correlate clinical and pathologic findings to establish a diagnosis.
AuthorshipAll the authors contributed equally to this work, regardless of the order of their surnames.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Aróstegui Aguilar J, Diago A, Carrillo Gijón R, Fernández Figueras M, Fraga J, García Herrera A, et al. Granulomas en dermatopatología: principales entidades. Parte I. Actas Dermosifiliogr. 2021. https://doi.org/10.1016/j.ad.2021.04.002
This article is an initiative of the Dermopathology Research Group of the Spanish Academy of Dermatology and Venereology (AEDV) and the Spanish Pathology Society (SEAP).


www.publicationethics.org.
