

Las epidermólisis ampollosas hereditarias (EA) son un grupo heterogéneo de genodermatosis caracterizadas por fragilidad cutánea de tipo mecánico1,2.
Se realizó un estudio descriptivo y retrospectivo que incluyó a los pacientes diagnosticados de EA mediante estudio genético en el Hospital Clínico Universitario de Valencia, entre 1968 y 2018. No se disponía de ningún registro con los casos valorados sin diagnóstico molecular. En 2018, la población adscrita al Departamento de Salud Clínico-La Malvarrosa era de 344.019 personas3.
Las variables analizadas fueron: edad al diagnóstico, edad actual, antecedentes familiares de EA, manifestaciones clínicas, localización de las lesiones, plano de separación dermo-epidérmico, estudio histológico, gen mutado, mutación, tipo de herencia, cigosidad, tipo de EA, tratamiento y complicaciones (tabla 1). Respecto a las complicaciones, se evaluó la presencia de caries, erosiones y úlceras orales, reflujo gastroesofágico, estreñimiento, retraso del crecimiento, anemia, enfermedad renal, sindactalia, carcinoma epidermoide cutáneo (CEC) y depresión.
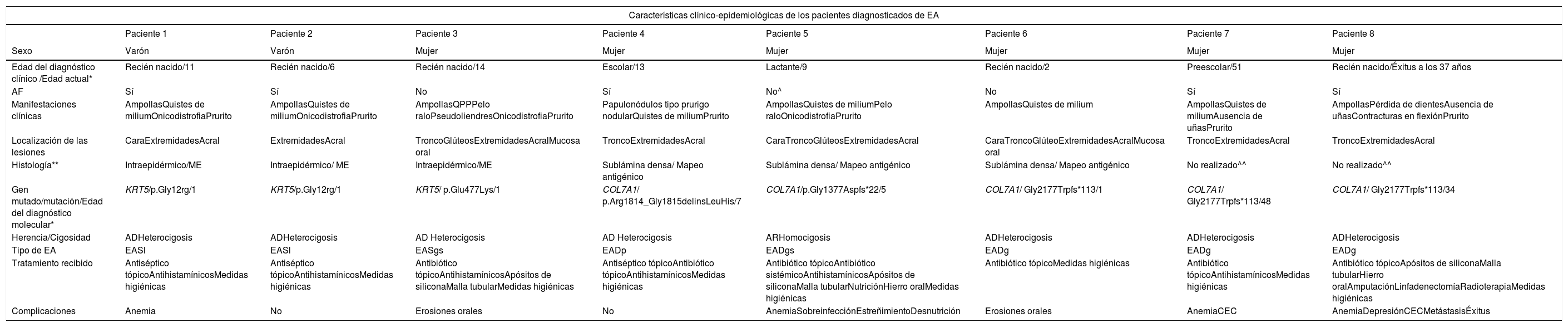
| Características clínico-epidemiológicas de los pacientes diagnosticados de EA | ||||||||
|---|---|---|---|---|---|---|---|---|
| Paciente 1 | Paciente 2 | Paciente 3 | Paciente 4 | Paciente 5 | Paciente 6 | Paciente 7 | Paciente 8 | |
| Sexo | Varón | Varón | Mujer | Mujer | Mujer | Mujer | Mujer | Mujer |
| Edad del diagnóstico clínico /Edad actual* | Recién nacido/11 | Recién nacido/6 | Recién nacido/14 | Escolar/13 | Lactante/9 | Recién nacido/2 | Preescolar/51 | Recién nacido/Éxitus a los 37 años |
| AF | Sí | Sí | No | Sí | No^ | No | Sí | Sí |
| Manifestaciones clínicas | AmpollasQuistes de miliumOnicodistrofiaPrurito | AmpollasQuistes de miliumOnicodistrofiaPrurito | AmpollasQPPPelo raloPseudoliendresOnicodistrofiaPrurito | Papulonódulos tipo prurigo nodularQuistes de miliumPrurito | AmpollasQuistes de miliumPelo raloOnicodistrofiaPrurito | AmpollasQuistes de milium | AmpollasQuistes de miliumAusencia de uñasPrurito | AmpollasPérdida de dientesAusencia de uñasContracturas en flexiónPrurito |
| Localización de las lesiones | CaraExtremidadesAcral | ExtremidadesAcral | TroncoGlúteosExtremidadesAcralMucosa oral | TroncoExtremidadesAcral | CaraTroncoGlúteosExtremidadesAcral | CaraTroncoGlúteoExtremidadesAcralMucosa oral | TroncoExtremidadesAcral | TroncoExtremidadesAcral |
| Histología** | Intraepidérmico/ME | Intraepidérmico/ ME | Intraepidérmico/ME | Sublámina densa/ Mapeo antigénico | Sublámina densa/ Mapeo antigénico | Sublámina densa/ Mapeo antigénico | No realizado^^ | No realizado^^ |
| Gen mutado/mutación/Edad del diagnóstico molecular* | KRT5/p.Gly12rg/1 | KRT5/p.Gly12rg/1 | KRT5/ p.Glu477Lys/1 | COL7A1/ p.Arg1814_Gly1815delinsLeuHis/7 | COL7A1/p.Gly1377Aspfs*22/5 | COL7A1/ Gly2177Trpfs*113/1 | COL7A1/ Gly2177Trpfs*113/48 | COL7A1/ Gly2177Trpfs*113/34 |
| Herencia/Cigosidad | ADHeterocigosis | ADHeterocigosis | AD Heterocigosis | AD Heterocigosis | ARHomocigosis | ADHeterocigosis | ADHeterocigosis | ADHeterocigosis |
| Tipo de EA | EASl | EASl | EASgs | EADp | EADgs | EADg | EADg | EADg |
| Tratamiento recibido | Antiséptico tópicoAntihistamínicosMedidas higiénicas | Antiséptico tópicoAntihistamínicosMedidas higiénicas | Antibiótico tópicoAntihistamínicosApósitos de siliconaMalla tubularMedidas higiénicas | Antiséptico tópicoAntibiótico tópicoAntihistamínicosMedidas higiénicas | Antibiótico tópicoAntibiótico sistémicoAntihistamínicosApósitos de siliconaMalla tubularNutriciónHierro oralMedidas higiénicas | Antibiótico tópicoMedidas higiénicas | Antibiótico tópicoAntihistamínicosMedidas higiénicas | Antibiótico tópicoApósitos de siliconaMalla tubularHierro oralAmputaciónLinfadenectomíaRadioterapiaMedidas higiénicas |
| Complicaciones | Anemia | No | Erosiones orales | No | AnemiaSobreinfecciónEstreñimientoDesnutrición | Erosiones orales | AnemiaCEC | AnemiaDepresiónCECMetástasisÉxitus |
Plano de separación en la unión dermo-epidérmica y estudio histológico realizado (ME: microscopía electrónica o mapeo antigénico);^^Se solicitó el estudio genético directamente; AF: antecedentes familiares; QPP: queratodermia palmo-plantar; KRT5: gen de la queratina 5; COL7A1: gen del colágeno 7; AD: autosómico dominante; AR: autosómico recesivo; EASl: epidermólisis ampollosa simple localizada; EASgs: epidermólisis ampollosa simple generalizada severa; EADp: epidermólisis ampollosa distrófica pruriginosa; EADgs: epidermólisis ampollosa distrófica generalizada severa; EADg: epidermólisis ampollosa distrófica generalizada; CEC: carcinoma epidermoide cutáneo.
El número de pacientes fue de ocho; tres se diagnosticaron de EA simple y cinco de EA distrófica. Los subtipos más frecuentes fueron la EA simple localizada y la EA distrófica generalizada. El 75% eran mujeres, el 62,5% se diagnosticaron al nacimiento y presentaban antecedentes familiares. Los pacientes 1 y 2, y el 7 y 8 eran hermanos. La manifestación clínica más frecuente fueron las ampollas (87,5%) y se localizaron principalmente en las extremidades. Todos los pacientes recibieron tratamiento tópico y seis precisaron hidroxicina como tratamiento sintomático del prurito. El tratamiento tópico consistió en antibióticos, curas locales con antisépticos y apósitos primarios (apósitos de silicona) y/o con apósitos secundarios (apósitos algodonosos y/o malla tubular). A todos los pacientes se les indicaron medidas higiénicas, que incluyeron: usar ropa de algodón, realizar duchas sin jabón, usar loción hidratante, secar el cuerpo con toques delicados, drenar el contenido de las ampollas con aguja estéril sin desepitelizarlas y usar clorhexidina 0,5% como antiséptico. La paciente con EA distrófica (EAD) generalizada severa requirió gabapentina por prurito refractario y soporte nutricional por desnutrición. Una paciente con EAD generalizada presentó un CEC metastásico en la pierna derecha que se trató con amputación, linfadenectomía inguinal y radioterapia paliativa. La anemia fue la complicación más frecuente (50%) y dos pacientes requirieron tratamiento con hierro oral por presentar una cifra de hemoglobina menor a 10g/dL. La evolución clínica fue variable, desde la mejoría de las lesiones hasta el fallecimiento de una paciente por un CEC metastásico. (fig. 1)
 Ampolla en la palma de la mano; 1B) Onicodistrofia; 1C) Cicatrización con quistes de milium en zonas sometidas a traumatismos. Paciente 3: Recién nacida con EASgs; 1D) Ampollas y erosiones cutáneas generalizadas; Control en la infancia; 1E, 1H) Ampollas de distribución herpetiforme en la región lumbar, glútea y muslos junto con signos de cicatrización atrófica; Control en la adolescencia; 1F) Onicodistrofia; 1G, 1I) Queratodermia palmoplantar; 1J) Pseudoliendres o vainas peripilares. Paciente 4: Niña de 7 años con EADp; 1K, 1L, 1M) Papulonódulos pruriginosos localizados en las superficies de extensión de las extremidades.")
Paciente 1: Niño de 10 años con EASl; 1A) Ampolla en la palma de la mano; 1B) Onicodistrofia; 1C) Cicatrización con quistes de milium en zonas sometidas a traumatismos. Paciente 3: Recién nacida con EASgs; 1D) Ampollas y erosiones cutáneas generalizadas; Control en la infancia; 1E, 1H) Ampollas de distribución herpetiforme en la región lumbar, glútea y muslos junto con signos de cicatrización atrófica; Control en la adolescencia; 1F) Onicodistrofia; 1G, 1I) Queratodermia palmoplantar; 1J) Pseudoliendres o vainas peripilares. Paciente 4: Niña de 7 años con EADp; 1K, 1L, 1M) Papulonódulos pruriginosos localizados en las superficies de extensión de las extremidades.
Las EA se producen por mutaciones en los genes que codifican las proteínas estructurales de la unión dermoepidérmica. El nivel de separación histopatológico y la mutación genética define el tipo y subtipo de EA. Los tipos de EA son: EA simple (EAS), EA juntural (EAJ), EA distrófica (EAD) y síndrome de Kindler1,2,4. En la EAS la ampolla aparece en el plano epidérmico, en la EAJ en la lámina lúcida y en la EAD en la sublámina densa. La EAJ y la EAD suelen ser más graves porque pueden afectar a otros órganos cubiertos por un epitelio4–6.
La EAS suele presentarse en el periodo neonatal. Las cicatrices, los quistes de milium y la distrofia ungueal son menos frecuentes que en la EAJ y EAD. La EAS localizada es el subtipo más frecuente y se presenta con ampollas en las palmas y plantas. La EAS generalizada severa se caracteriza por ampollas generalizadas en el periodo neonatal, distribución herpetiforme de las lesiones en la infancia y desarrollo de una queratodermia palmoplantar1,2,4.
La característica más constante de la EAJ es la hipoplasia del esmalte, y en las formas graves, la aparición de un tejido de granulación exuberante en las regiones periorificiales y los pliegues1,2,4. Las EAD autosómico recesivas son las más graves causando una enfermedad ampollosa, erosiva y mutilante con compromiso cutáneo-mucoso y de los órganos internos. El CEC es más frecuente y agresivo en los pacientes con EAD, considerándose como la principal causa de muerte en la EA1,2,4–7. En contraste con la experiencia de Feinstein et al.8, en esta serie destaca el predominio de las formas de EAD autosómica dominante frente a las formas recesivas. Además, es interesante remarcar el desarrollo de CEC en las dos mujeres adultas con EAD generalizada.
En el proceso diagnóstico se debe realizar una biopsia cutánea de una lesión inducida por fricción para realizar un mapeo antigénico histológico que establecerá el plano de separación dermo-epidérmica y orientará el tipo de EA y el estudio genético1,2. El diagnóstico molecular es fundamental, ya que aporta información esencial para el pronóstico y para un manejo adecuado de los pacientes, y proporciona un consejo genético a los padres e individuos afectados2,4.
El tratamiento es sintomático y multidisciplinar, encontrándose en desarrollo terapias proteicas, celulares y génicas para la EAD1,2,4,9.
En conclusión, la prevalencia de EA en nuestro departamento de salud es similar a la descrita en estudios previos10. La mayoría de los pacientes eran mujeres y se diagnosticaron al nacimiento. Las manifestaciones clínicas fueron heterogéneas, siendo las ampollas, la onicodistrofia y el prurito las más frecuentes. La complicación más frecuente fue la anemia, lo cual puede estar relacionado con la mayor frecuencia de EAD en nuestra serie. El tratamiento, principalmente, fue sintomático.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



