

Hereditary epidermolysis bullosa (EB) comprises a heterogeneous group of genodermatoses characterized by mechanical skin fragility.1,2
We conducted a descriptive, retrospective study of all patients genetically diagnosed with EB at Hospital Clínico Universitario de Valencia between 1968 and 2018. There were no cases on record that had been assessed without a molecular diagnosis. In 2018, the corresponding health district, La Malvarrosa, offered coverage to a population of 344 019 people.3
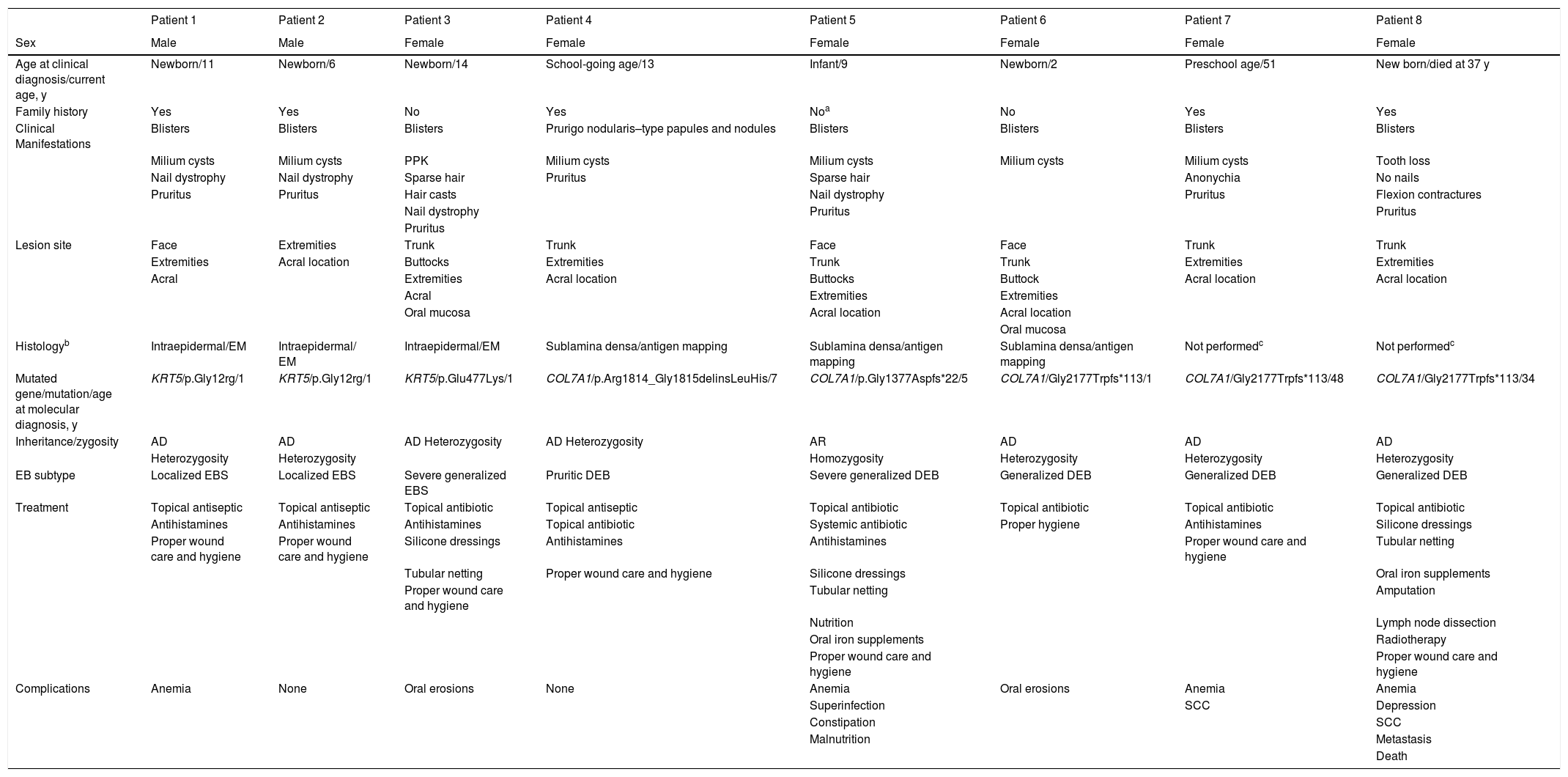
The following variables were analyzed: age at diagnosis, current age, family history of EB, clinical manifestations, location of lesions, dermoepidermal cleavage plane, histologic features, mutated gene, mutation, mode of inheritance, zygosity, type of EB, treatment, and complications (Table 1). Presence of the following complications was assessed: oral cavities, oral erosions and ulcers, gastroesophageal reflux, constipation, growth retardation, anemia, kidney disease, syndactyly, cutaneous squamous cell carcinoma (SCC), and depression.
Clinical and epidemiological characteristics of patients with EB.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | |
|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Female | Female | Female | Female | Female |
| Age at clinical diagnosis/current age, y | Newborn/11 | Newborn/6 | Newborn/14 | School-going age/13 | Infant/9 | Newborn/2 | Preschool age/51 | New born/died at 37 y |
| Family history | Yes | Yes | No | Yes | Noa | No | Yes | Yes |
| Clinical Manifestations | Blisters | Blisters | Blisters | Prurigo nodularis–type papules and nodules | Blisters | Blisters | Blisters | Blisters |
| Milium cysts | Milium cysts | PPK | Milium cysts | Milium cysts | Milium cysts | Milium cysts | Tooth loss | |
| Nail dystrophy | Nail dystrophy | Sparse hair | Pruritus | Sparse hair | Anonychia | No nails | ||
| Pruritus | Pruritus | Hair casts | Nail dystrophy | Pruritus | Flexion contractures | |||
| Nail dystrophy | Pruritus | Pruritus | ||||||
| Pruritus | ||||||||
| Lesion site | Face | Extremities | Trunk | Trunk | Face | Face | Trunk | Trunk |
| Extremities | Acral location | Buttocks | Extremities | Trunk | Trunk | Extremities | Extremities | |
| Acral | Extremities | Acral location | Buttocks | Buttock | Acral location | Acral location | ||
| Acral | Extremities | Extremities | ||||||
| Oral mucosa | Acral location | Acral location | ||||||
| Oral mucosa | ||||||||
| Histologyb | Intraepidermal/EM | Intraepidermal/ EM | Intraepidermal/EM | Sublamina densa/antigen mapping | Sublamina densa/antigen mapping | Sublamina densa/antigen mapping | Not performedc | Not performedc |
| Mutated gene/mutation/age at molecular diagnosis, y | KRT5/p.Gly12rg/1 | KRT5/p.Gly12rg/1 | KRT5/p.Glu477Lys/1 | COL7A1/p.Arg1814_Gly1815delinsLeuHis/7 | COL7A1/p.Gly1377Aspfs*22/5 | COL7A1/Gly2177Trpfs*113/1 | COL7A1/Gly2177Trpfs*113/48 | COL7A1/Gly2177Trpfs*113/34 |
| Inheritance/zygosity | AD | AD | AD Heterozygosity | AD Heterozygosity | AR | AD | AD | AD |
| Heterozygosity | Heterozygosity | Homozygosity | Heterozygosity | Heterozygosity | Heterozygosity | |||
| EB subtype | Localized EBS | Localized EBS | Severe generalized EBS | Pruritic DEB | Severe generalized DEB | Generalized DEB | Generalized DEB | Generalized DEB |
| Treatment | Topical antiseptic | Topical antiseptic | Topical antibiotic | Topical antiseptic | Topical antibiotic | Topical antibiotic | Topical antibiotic | Topical antibiotic |
| Antihistamines | Antihistamines | Antihistamines | Topical antibiotic | Systemic antibiotic | Proper hygiene | Antihistamines | Silicone dressings | |
| Proper wound care and hygiene | Proper wound care and hygiene | Silicone dressings | Antihistamines | Antihistamines | Proper wound care and hygiene | Tubular netting | ||
| Tubular netting | Proper wound care and hygiene | Silicone dressings | Oral iron supplements | |||||
| Proper wound care and hygiene | Tubular netting | Amputation | ||||||
| Nutrition | Lymph node dissection | |||||||
| Oral iron supplements | Radiotherapy | |||||||
| Proper wound care and hygiene | Proper wound care and hygiene | |||||||
| Complications | Anemia | None | Oral erosions | None | Anemia | Oral erosions | Anemia | Anemia |
| Superinfection | SCC | Depression | ||||||
| Constipation | SCC | |||||||
| Malnutrition | Metastasis | |||||||
| Death |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; COL7A1, collagen 7 gene; DEB, Dystrophic epydermolysis bullosa; EB, epidermolysis bullosa; EBS, epidermolysis bullosa simplex; EM, electron microscopy; KRT5, keratin 5 gene; PPK, palmoplantar keratoderma; SSC, squamous cell carcinoma.
Eight patients diagnosed with EB during the study period were identified: 3 had EB simplex (EBS) and 5 had dystrophic EB (DEB). The most common subtypes were localized EBS and generalized DEB; 6 of the patients (75%) were women and 5 (62.5%) had been diagnosed at birth and had a family history of EB. Patients 1 and 2 and 7 and 8 were siblings. Blisters were the most common clinical manifestation (87.5%) and were mainly located on the extremities. All patients received topical treatments and 6 required symptomatic treatment with hydroxyzine for pruritus. Topical treatments consisted of antibiotics, local therapy with antiseptics and primary dressings (silicone), and/or secondary dressings (cotton dressings with or without tubular netting). All the patients were instructed on proper wound care and hygiene and were advised to wear cotton fabrics, shower without soap, apply moisturizing creams, pat their body dry, drain their blisters with a sterile needle without breaking the skin, and apply antiseptic chlorhexidine 0.5%. The patient with severe generalized DEB required gabapentin for refractory pruritus and nutritional support for malnutrition. One patient with generalized DEB developed metastatic SCC on her right leg. She was treated with amputation, inguinal lymph node dissection, and palliative radiotherapy. Anemia was the most common complication (50%) and 2 patients with a hemoglobin level of less than 10 g/dL required oral iron supplementation. The clinical course was variable and ranged from improvement of lesions to death due to metastatic SCC (Fig. 1).
. Patient 4: 7-year-old girl with pruritic dystrophic epidermolysis bullosa. K,L,M, Pruritic papules and nodules on the surface of the extremities.")
Patient 1: 10-year-old boy with localized epidermolysis bullosa simplex. A, Blister on his palm. B, Nail dystrophy. C, Scarring with milium cysts at sites of trauma. Patient 3: Newborn with severe generalized epidermolysis bullosa simplex. D, Generalized skin blisters and erosions; controlled in childhood. E, H, Herpetiform blistering in lumbar, buttock, and thigh areas together with signs of atrophic scarring; controlled in adolescence. F, Nail dystrophy. G,I, Palmoplantar keratoderma; J, Hair casts (pseudonits). Patient 4: 7-year-old girl with pruritic dystrophic epidermolysis bullosa. K,L,M, Pruritic papules and nodules on the surface of the extremities.
EB is caused by mutations in genes encoding the structural proteins in the dermoepidermal junction. EB type and subtype are defined by the mutation involved and the level of skin cleavage on histology. There are 4 types of EB: EBS, junctional EB (JEB), DEB, and Kindler syndrome.1,2,4 Blisters form in the epidermis in EBS, the lamina lucida in JEB, and the sublamina densa in DEB. JEB and DEB are usually more severe as they can affect other organs with an epithelial lining.4–6
EBS usually manifests in the neonatal period. Scars, milium cysts, and nail dystrophy are less common than in JEB or DEB. The most common subtype of EB is localized EBS, which is characterized by palmoplantar blisters. Severe generalized EBS is characterized by generalized blistering in the neonatal period, herpetiform blistering in childhood, and palmoplantar keratoderma.1,2,4
Enamel hypoplasia is the most consistent form of JEB and patients with severe disease can develop exuberant granulation tissue in the periorificial regions and skin folds.1,2,4 Autosomal recessive DEB is the most severe subtype of EB and causes bullous, erosive, mutilating disease with involvement of the skin, mucous membranes, and internal organs. SSC is the main cause of death in EB and is more common—and aggressive—in patients with dystrophic forms.1,2,4–7 Contrasting with findings by Feinstein et al.,8 autosomal dominant DEB was more common than recessive DEB in our series. Also of note in our series was the presence of SCC in the 2 women with generalized DEB.
The diagnostic work-up in EB should include skin biopsy of a friction-induced lesion to enable immunofluorescence antigen mapping, which will show the level of cleavage in the dermoepidermal junction, helping to classify the type of EB and guide molecular testing.1,2 Molecular diagnosis is essential, as it provides key prognostic information and guides management. It also enables clinicians to offer genetic counseling to patients and parents of affected individuals.2,4
Treatment is symptomatic and multidisciplinary, and several protein-, cell-, and gene-based therapies are currently under development for DEB.1,2,4,9
In conclusion, the prevalence of AD in our health care setting is similar to that described in previous studies.10 Most of the patients were female and had been diagnosed at birth. The clinical manifestations were variable, but the most common findings were blisters, nail dystrophy, and pruritus. The main complication was anemia, possibly because of the higher prevalence of DEB. Treatment was largely symptomatic.
FundingNo funding was received for this study.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Guillen-Climent S, Fernández García L, García-Vázquez A, Martín JM. Epidermólisis ampollosa hereditaria: serie de casos. Actas Dermosifiliogr. 2021;112:954–958.


