


The ectodermal dysplasias are a large group of hereditary disorders characterized by alterations of structures of ectodermal origin. Although some syndromes can have specific features, many of them share common clinical characteristics. Two main groups of ectodermal dysplasias can be distinguished. One group is characterized by aplasia or hypoplasia of ectodermal tissues, which fail to develop and differentiate because of a lack of reciprocal signaling between ectoderm and mesoderm, the other has palmoplantar keratoderma as its most striking feature, with additional manifestations when other highly specialized epithelia are also involved. In recent decades, the genes responsible for at least 30 different types of ectodermal dysplasia have been identified, throwing light on the pathogenic mechanisms involved and their correlation with clinical findings.
Las displasias ectodérmicas son un amplio grupo de trastornos hereditarios que se caracterizan por la alteración de estructuras derivadas del ectodermo. Aunque algunos de estos síndromes poseen características específicas, determinados rasgos clínicos son comunes en muchos de ellos. De modo general, se diferencian 2 grupos de trastornos: uno caracterizado por la aplasia o hipoplasia de los derivados ectodérmicos, que fracasan en su desarrollo y diferenciación por la ausencia de señales recíprocas específicas entre ectodermo y mesénquima, y otro en el que la característica más llamativa es la queratodermia palmoplantar, que se presenta en asociación con otras manifestaciones cuando se afectan otros epitelios altamente especializados. En las últimas décadas se ha logrado identificar el gen responsable en al menos 30 entidades, permitiéndonos entender los mecanismos patogénicos y su correlación con la clínica.
The ectoderm is one of the primitive embryonic components. At around the third week of development, it undergoes a subdivision into the neuroectoderm, the origin of the nervous system, and the ectoderm, which will envelop the entire embryonic surface and form the epidermis, epidermal appendages, and tooth enamel. The ectoderm therefore gives rise not only to hair, teeth, nails, and sweat glands, but also to the central nervous system, peripheral nervous system, eyes, ears, and nose, as well as the eccrine, mammary, and pituitary glands.1 During development, the ectoderm undergoes complex interactions with the mesoderm, so ectodermal disorders may lead to abnormalities in mesodermal structures such as the musculoskeletal and genitourinary systems.2
Ectodermal dysplasias (EDs) are a heterogenous group of hereditary disorders characterized by certain shared structural and functional abnormalities in tissues derived from the ectoderm. Most of these diseases are also associated with an abnormal development of structures derived from the mesoderm and, occasionally, mental retardation. They are considered rare conditions, with an estimated incidence of 7 cases per 10 000 births.3 They can be transmitted by any of the possible Mendelian inheritance patterns,4 and although many share certain clinical characteristics, some syndromes have specific clinical findings. To date, approximately 200 such conditions are known, and the causative gene mutation has been identified in around 30. Mutations in only 4 genes (EDA1, EDAR, EDARADD, and WNT10A) are responsible for most cases of ED.5
Historical PerspectiveThe first descriptions of clinical cases that might correspond to what we would now classify as ED date from 1792.6 In 1848, Thurman defined anhidrotic ectodermal dysplasia (also known as hypohidrotic ectodermal dysplasia [HED]) as a condition in its own right.7 Subsequently, similar cases were reported, such as the one presented by Weddernhorn and published in 1875 by the naturist Charles Darwin: “I may give an analogous case, communicated to me by Mr. W. Weddenburn, of a Hindoo family in Scinde, in which ten men, in the course of four generations, were furnished, in both jaws taken together, with only four small and weak incisor teeth and with eight posterior molars. The men thus affected have very little hair on the body, and become bald early in life. They also suffer much during hot weather from excessive dryness of the skin. It is remarkable that no instance has occurred of a daughter being affected…though the daughters in the above family are never affected, they transmit the tendency to their sons: and no case has occurred of a son transmitting it to his sons. The affection thus appears only in alternate generations, or after long intervals.”8 The above case described by Darwin corresponds to what we would describe today as X-linked HED, a term coined by Weech in 1929.9
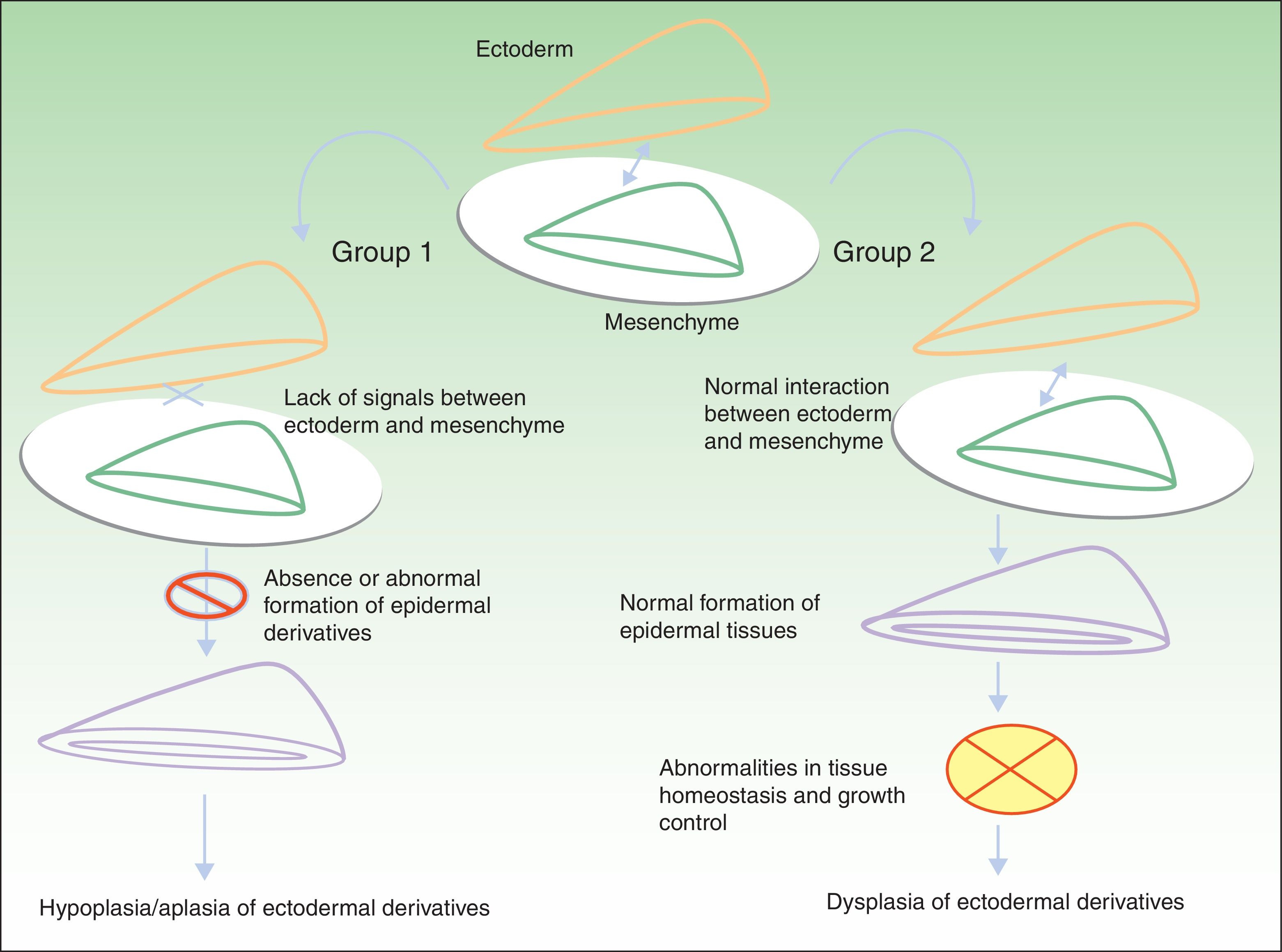
Classification of Ectodermal DysplasiasThe classification of EDs is complex, and classification systems have come and gone in an attempt to accommodate clinical and genetic data.10–15 Biomolecular findings have enabled the identification of the causative mutations that become manifest through 2 broad pathogenic mechanisms, associated with specific clinical features. Using these mechanisms as a starting point, in 2009, Priolo2 established a clinical-functional classification, which will form the basis for this review. The author proposed the definition of 2 groups of disorders (Fig. 1).

Pathogenic mechanisms in ectodermal dysplasias. In group 1, there is an abnormal interaction between the ectoderm and the mesenchyme, thereby impeding correct differentiation of the epidermal derivatives, which are hypoplastic or aplastic. In group 2, the interaction between the ectoderm and mesenchyme is normal, and the epidermal derivatives differentiate normally, but tissue homeostasis and growth are abnormal, so dysplasia is present in the ectodermal derivatives. Adapted from Priolo.2
Group 1 corresponds to disorders in which defective interaction between the ectoderm and the mesenchyme is apparent. Two pathophysiologic mechanisms have been identified:
- 1.
Changes in the signaling pathways that modulate activity of nuclear factor (NF) κB (ectodysplasin/ectodysplasin-A [EDA] receptor [EDAR]/EDAR associated death domain [EDARADD] signaling pathway and NEMO [NF-κB essential modulator] regulatory pathway).
- 2.
Regulatory changes in transcription and/or expression of genes such as p63, DLX3, MSX1, EVC2, and EVC.
The resulting clinical phenotype is hypoplasia or aplasia of structures derived from the ectoderm. Development and differentiation of these structures fail due to the absence of specific reciprocal signals between the ectoderm and the mesenchyme (Table 1).
Group 1 Disorders.
| Changes in the Signaling Pathways that Modulate NF-κB Activity | ||||||
|---|---|---|---|---|---|---|
| Pathway | Gene | Locus | Protein | Disease | OMIM | Type of Inheritance |
| Ectodysplasin-EDAR-EDARRADD signaling pathway | ED-1 | Xq12-q13 | Ectodysplasin | Anhidrotic ectodermal dysplasia | omim:305100305100 | XL |
| EDAR | 2q13 | EDAR | Anhidrotic ectodermal dysplasia | omim:129490129490 | AD | |
| Anhidrotic ectodermal dysplasia | omim:224900224900 | AR | ||||
| EDARADD | 1q42-2-q.43 | EDAR-associated death domain | Anhidrotic ectodermal dysplasia | omim:229400229400 | AR | |
| NEMO regulatory pathway | NEMO/IkKγ | Xq28 | NF-κB | Incontinentia pigmenti | omim:308300308300 | XL |
| Anhidrotic ectodermal dysplasia with immunodeficiency | omim:300291300291 | XL | ||||
| Anhidrotic ectodermal dysplasia with osteopetrosis and immunodeficiency | omim:300301300301 | XL | ||||
| IkBα | 14q13 | IkBα | Anhidrotic ectodermal dysplasia with immunodeficiency | omim:164008164008 | AD | |
| Abnormalities in Gene Transcription/Expression Regulators | |||||
|---|---|---|---|---|---|
| Gene | Locus | Protein | Disease | OMIM | Type of Inheritance |
| p63 | 3q27 | P63 | EEC syndrome | omim:604292604292 | AD |
| AEC syndrome | omim:106260106260 | AD | |||
| ADULT syndrome | omim:103285103285 | AD | |||
| Limb-mammary syndrome | omim:603543603543 | AD | |||
| Rapp-Hodgkin syndrome | omim:603543603543 | AD | |||
| DLX3 | 17q21 | DLX3 | Tricho-dento-osseus syndrome | omim:190320190320 | AD |
| MSX1 | 4p16.1 | MSX1 | Witkop disease | omim:189500189500 | AD |
| EVC2 | 4p16 | EVC2 | Ellis-van Creveld disease | omim:225500225500 | AR |
| EVC | 4p16 | EVC | Weyers acrodental dysostosis | omim:193530193530 | AD |
| Ellis-van Creveld disease | omim:225500225500 | AR | |||
Adapted from Priolo.2 Abbreviations: AD, autosomal dominant transmission; ADULT; acro-dermato-ungual-lacrimal-tooth; AEC, ankyloblepharon-ectodermal dysplasia-cleft lip/palate; AR, autosomal recessive transmission; EEC, ectrodactyly-ectodermal dysplasia-cleft lip/palate; OMIM, Online Mendelian Inheritance in Man; XL, X-linked.
Group 2 corresponds to disorders in which there is abnormal function of a structural protein in the cell membrane. Examples of structural proteins include nectin 1, connexins, and plakophilin, whose role in adhesion and cell-cell communication is essential for maintaining tissue homeostasis and controlling cell growth, development, and response to different stimuli.
Clinically, these disorders are mainly characterized by skin abnormalities such as palmoplantar keratoderma, with or without involvement of highly differentiated epithelia associated with deafness or retinal dystrophy (Table 2).
Group 2 Disorders.
| Gene | Locus | Protein | Disease | OMIM | Type of Transmission |
|---|---|---|---|---|---|
| GJB6 | 13q12 | Connexin 30 | Clouston syndrome | omim:129500129500 | AD |
| PVRL1 | 11q23-q24 | Nectin 1 | Ectodermal dysplasia with cleft lip/palate | omim:255060255060 | AD |
| PKP1 | 1q32 | Plakophyllin 1 | Ectodermal dysplasia with fragile skin syndrome | omim:604536604536 | AR |
| CDH3 | 16q22.1 | Cadherin 3 | Ectodermal dysplasia with ectrodactyly and macular dystrophy | omim:225280225280 | AR |
| WNT10A | 2q35 | Wnt10A | Odonto-onycho-dermal dysplasia | omim:257980257980 | AR |
Adapted from Priolo.2 Abbreviations: AD, autosomal dominant transmission; AR, autosomal recessive transmission; OMIM, Online Mendelian Inheritance in Man.
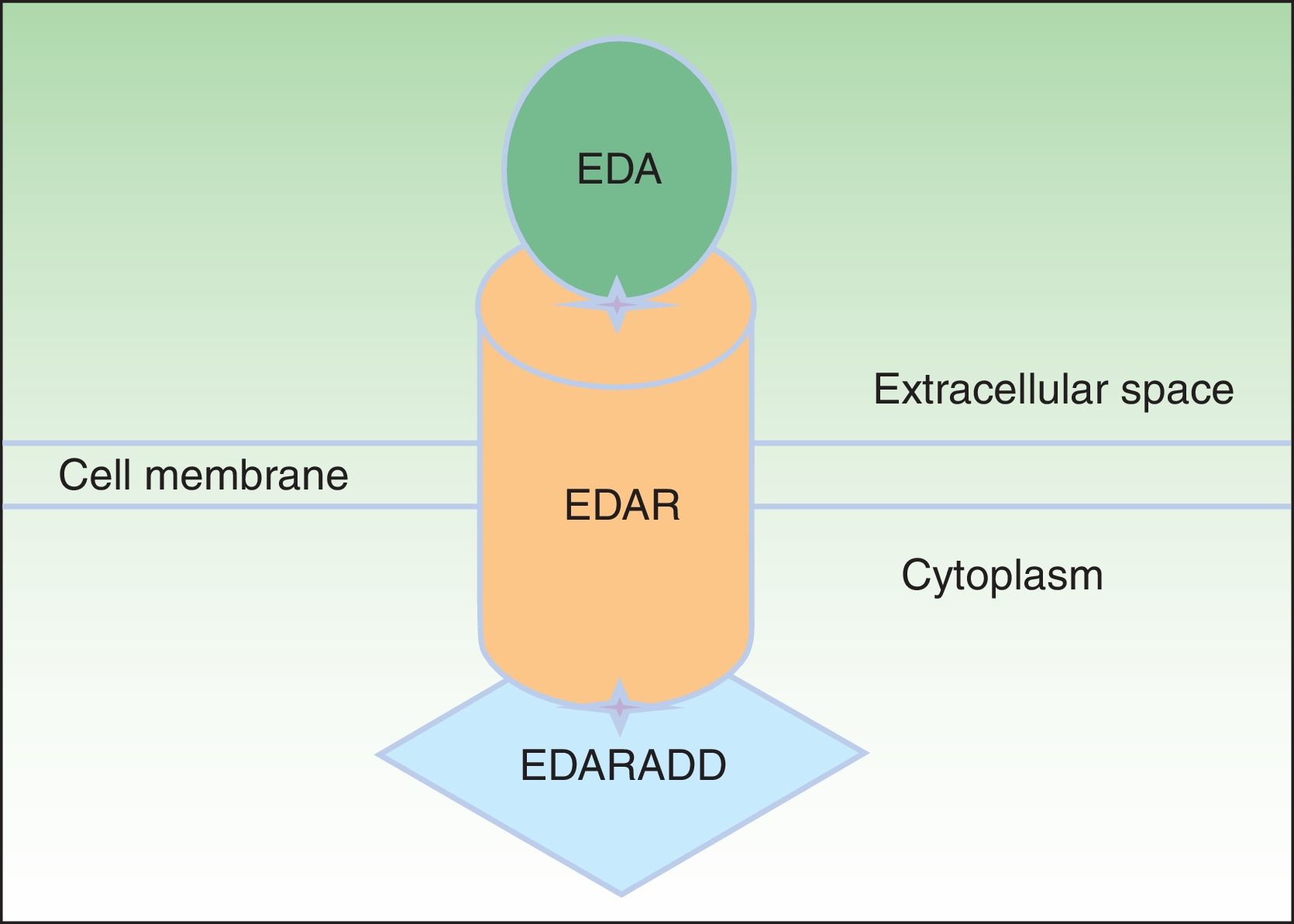
EDA, which consists of 391 amino acids, is a type 2 transmembrane protein belonging to the tumor necrosis factor (TNF) family.16 The most biologically important isoforms of EDA are EDA-A1 and EDA-A2.17 EDA-A1 binds to the EDA receptor (EDAR), whereas EDA-A2 binds to the X-linked EDAR A2 ligand (Fig. 2).18 EDAR is a type 1 transmembrane protein that consists of 448 amino acids and belongs to the TNF receptor superfamily. It has an extracellular region, a transmembrane region, and a death domain in its intracellular region.19 A death domain is a protein interaction module that interacts with the death domains of other proteins, thereby triggering metabolic cascades that are often implicated in regulating apoptosis and inflammation through the NF-κB cascade. The extracellular domain of EDAR is essential for binding to EDA-A1, whereas the death domain on the intracellular region plays an important role in the initiation of apoptotic transduction signals.20 The EDAR death domain has an associated death domain protein (EDARADD), which contains a 208 amino-acid sequence.21 The integrity of the death domains of both proteins is thus vital for their interaction22 and a normal regulation of embryonic morphogenesis.17 Therefore, the EDA-mediated signaling pathway is essential for appropriate organ development and ectoderm-derived structures, such as hair, nails, pituitary gland, mammary glands, sweat glands, nose, eyes, and tooth enamel.23
 protein binds to an ectodysplasin-A receptor (EDAR), located in the extracellular region of EDAR. EDAR has an extracellular region, a transmembrane region, and a death domain in its intracellular region. This death domain binds to the death domain of EDAR-associated death domain (EDARADD). Adapted from Lu et al.36")
Ectodysplasin-EDAR-EDARADD pathway. The ectodysplasin-A (EDA) protein binds to an ectodysplasin-A receptor (EDAR), located in the extracellular region of EDAR. EDAR has an extracellular region, a transmembrane region, and a death domain in its intracellular region. This death domain binds to the death domain of EDAR-associated death domain (EDARADD). Adapted from Lu et al.36
The EDAR gene, which has 12 exons, is located on chromosome 2 (locus 2q11-q13)19 and encodes the receptor for EDA-A1 (EDAR).17 To date, at least 41 EDAR mutations have been described. Most of these involve exon 12, which encodes the C-terminal region where the death domain is located.24 Some of these mutations have been reported in Spanish patients.25,26 The EDARADD gene, located on chromosome 1 (locus 1q-42-q43), encodes EDARADD, which is implicated in several diseases of autosomal recessive and dominant inheritance.17,27 Finally, certain mutations in the WNT10A gene, whose product is a member of the Wnt signaling pathway and implicated in embryonic development and cell differentiation, as well as in certain physiologic processes in adults and certain cancers, have been shown to give rise to several forms of ED of autosomal inheritance, such as HED or odonto-onycho-dermal dysplasia.5
X-linked HEDAlso known as Christ-Siemens-Touraine syndrome, X-linked HED is the most frequent form of ED, with an incidence of approximately 1 case per 100 000 births.28 Given the X-linked inheritance, affected males show all or most of the typical characteristics of the disease, while female carriers show only partial manifestations. There is no genotype-phenotype relationship, and the phenotype can vary greatly among different families and within the same family group.29 X-linked HED occurs as a result of mutations in the ED1 gene, also known as EDA.30 This gene is located on the long arm of the X chromosome (locus Xq12-q13) and encodes the EDA protein. More than 204 different ED1 mutations have been identified.24 Although a wide range of deletions and insertions have been reported, just 1 mutation accounts for 80% of the cases.31,32 Recently, a Spanish group has identified a family with a previously unreported mutation in this gene.33
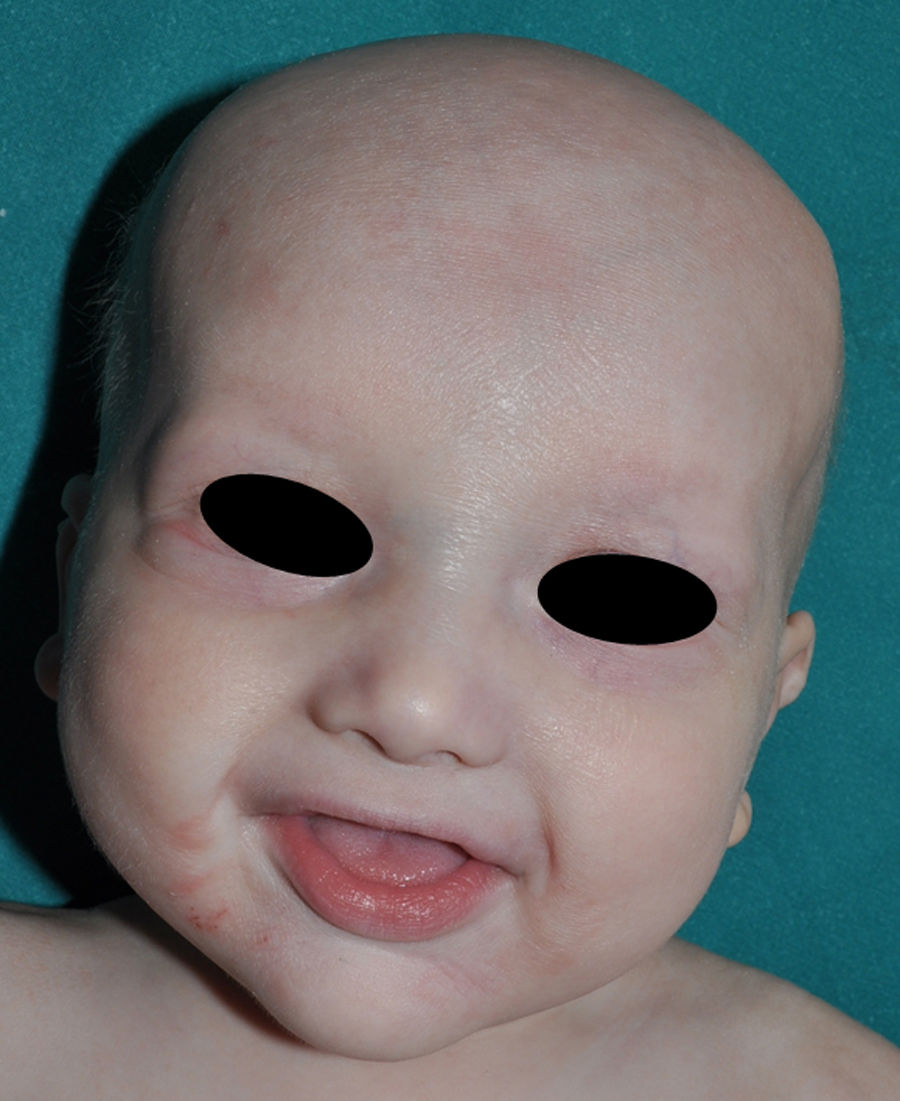
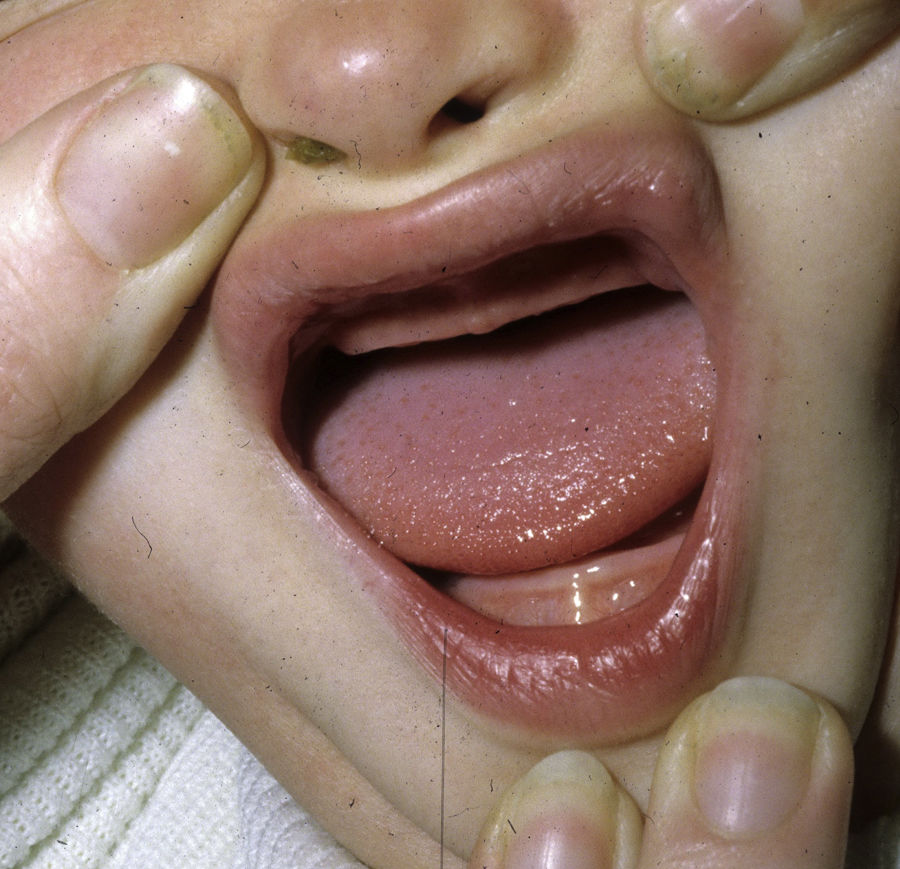
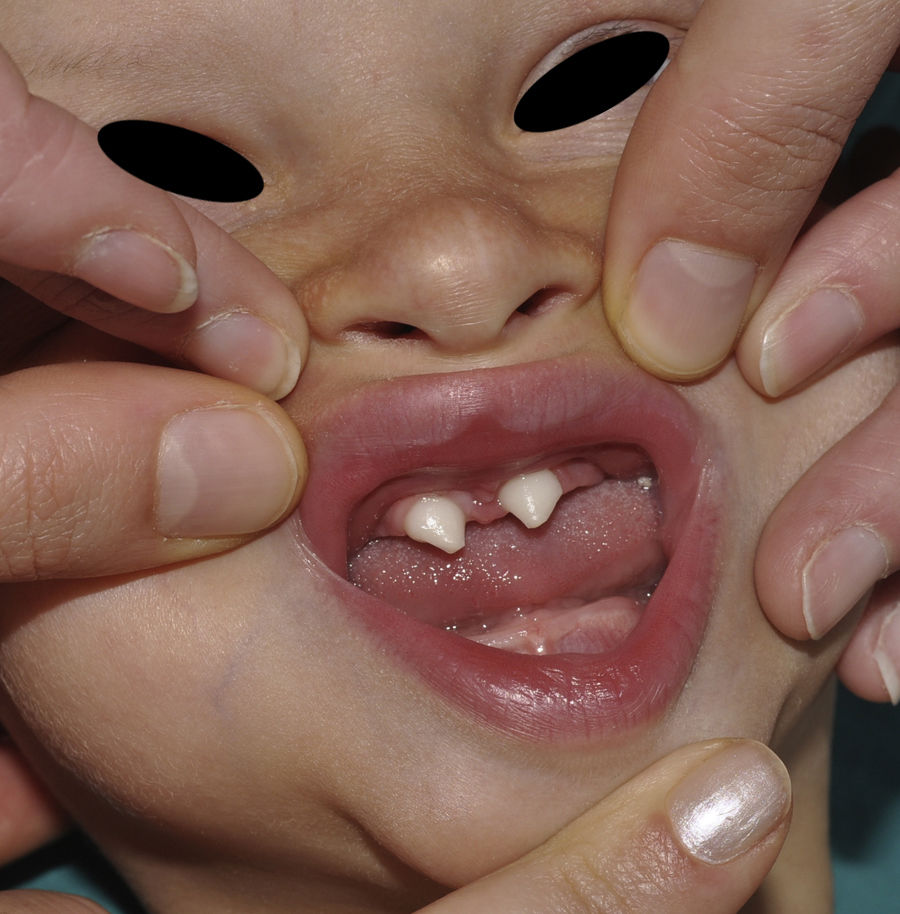
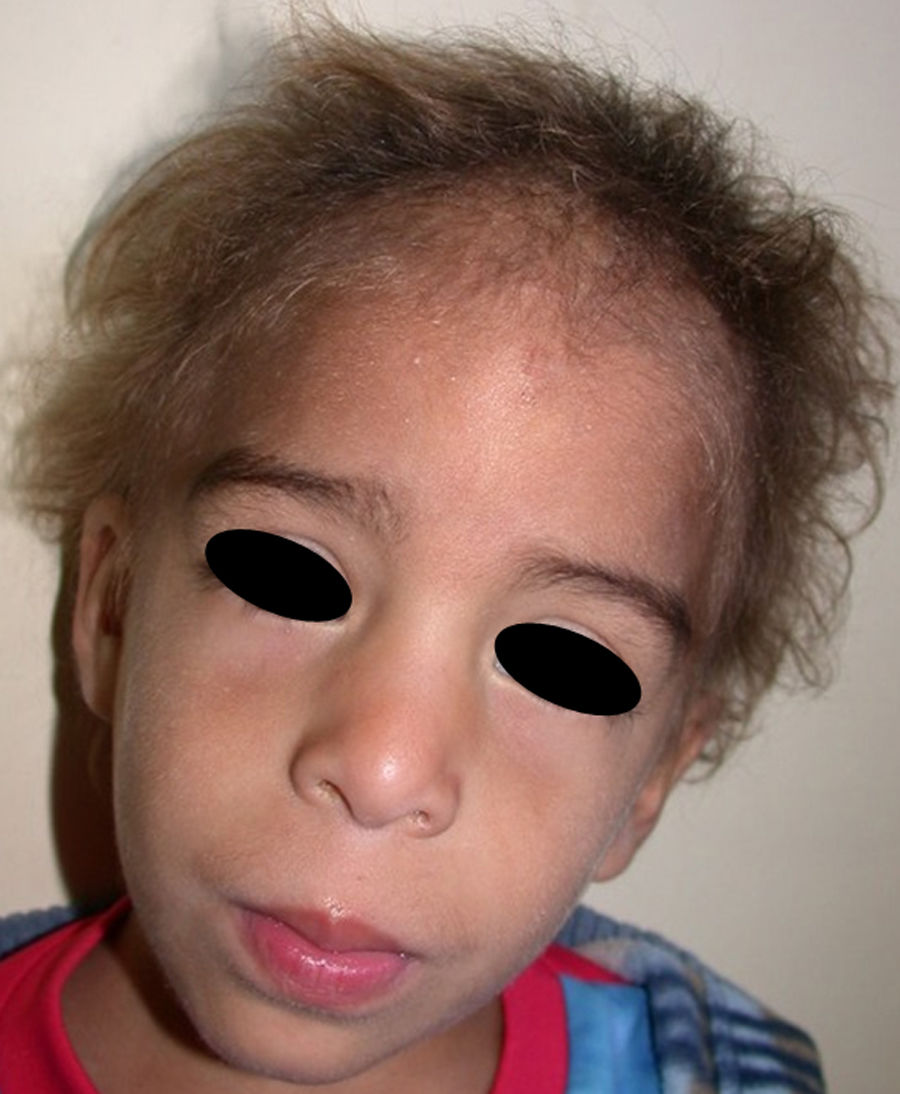
The distinctive clinical features include skin, tooth, and sweating abnormalities. In the neonate, the characteristic abnormalities are not especially notable, and so diagnosis in the first days of life is difficult. Up to 70% of boys with X-linked HED show skin desquamation during the neonatal period. This finding is also relatively common among female carriers. Cases of presentation as collodion baby have been reported.34 Alopecia is usually the first notable characteristic, although alopecia universalis is rare (Fig. 3).35 During childhood, most patients have sparse, fine, blond hair which darkens and thickens as the individuals get older. The eyebrows and beard are also sparse, but the eyelashes and armpit and pubic hair can be normal. Other body hair is also sparse or even absent.36 In some patients, hair shaft abnormalities may be present, though this finding is not specific for X-linked HED.37 Tooth abnormalities may become apparent during lactation as hypoplasia of the alveolar crests (Fig. 4). The number of missing and malformed teeth varies greatly between families and within the same family and there are also substantial variations between sexes.38 Morphologic variations are more evident in the anterior teeth. The most common type of affected tooth has an abnormal crown and takes on a cone or peg shape (Fig. 5).35 An X-ray will usually reveal taurodontism, an abnormality mainly of the molars characterized by a shortening of the root although the total height of the tooth is unchanged, yielding a prismatic form.38 The ability to sweat is reduced or absent, and so patients are predisposed to developing hyperthermia with physical exercise or high ambient temperatures.36 More than 90% of children have recurrent fever spikes with no apparent cause during the first year of life. Febrile seizures occur in approximately 6% of children with X-linked HED.39 Sweat disorders make a substantial contribution to the morbidity and mortality associated with X-linked HED; thus the mental retardation reported in between 30% and 50% of some series could be due to the damage caused by prolonged fever and febrile seizures.35 Although a genotype-phenotype correlation is apparent in terms of extent of sweat gland dysfunction, the risk of hyperthermia cannot be predicted and there is no correlation with morbidity and mortality.40



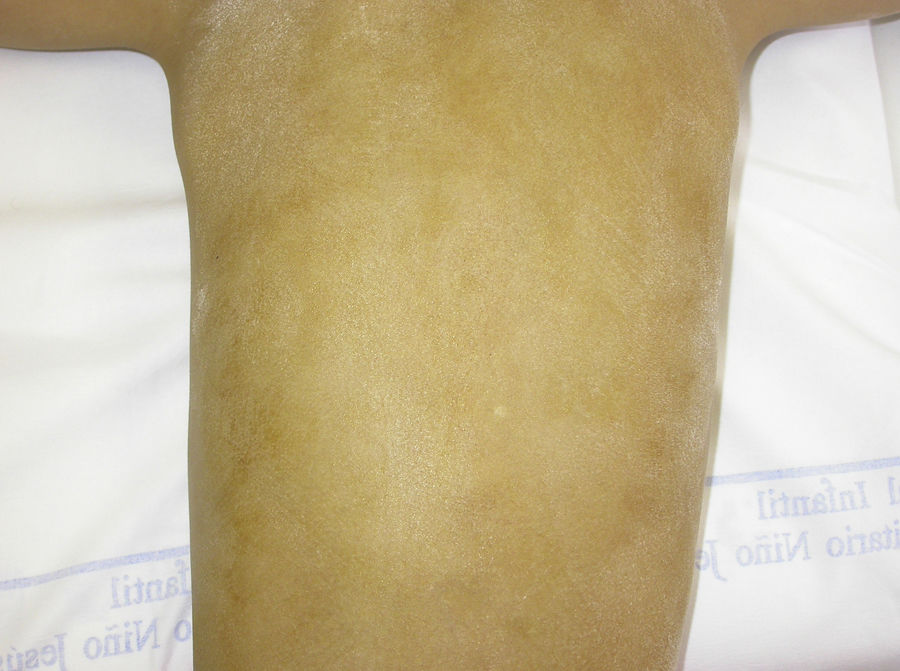
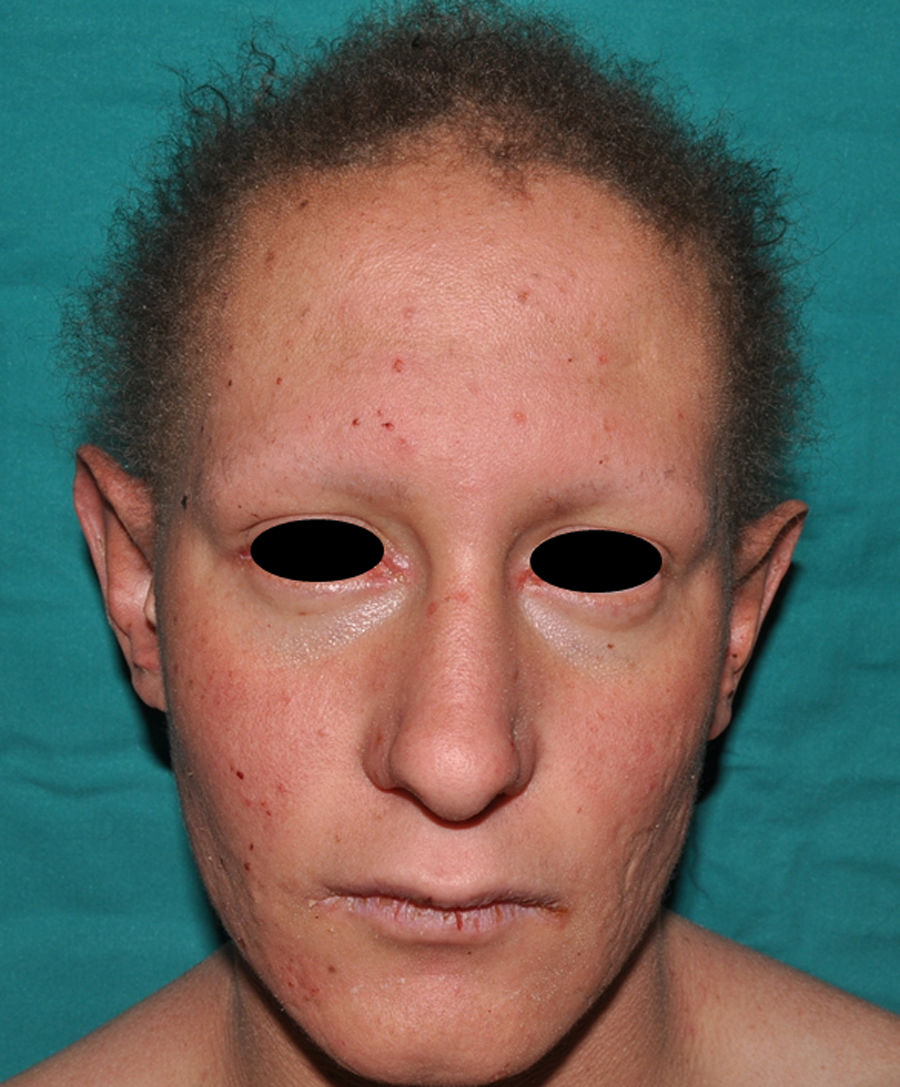
In the full syndrome, patients have characteristic facies, with a prominent frontal bone and chin, sunken nasal bridge, thick and everted lips, large ears, and a broad and high maxillary bone.35 Most abnormalities in craniofacial morphology can be attributed to the absence of teeth, although some authors have suggested that changes in embryonic morphogenesis could also be responsible.41 In addition to the typical characteristics, from childhood onwards, patients usually develop a dry, thin, shiny skin, periocular hyperpigmentation and fine periocular wrinkles giving the appearance of premature aging (Fig. 6), as well as small papular lesions reminiscent of sebaceous hyperplasia.35 Up to two-thirds of patients have atopic eczema, which can be difficult to control. Palmoplantar keratoderma is rare in X-linked HED, although it is often found in hidrotic ectodermal dysplasia and Clouston syndrome.42 The nails can be normal or fragile, but they are not usually especially dystrophic.35

Abnormalities in the mucosal glands can cause very thick nasal secretions that predispose patients to the development of respiratory tract infections.35,36 The decreased salivary secretion could, according to some authors, increase the risk of tooth decay and oral fungal infections, and also hinder food ingestion and speech.43 The reduction or absence of meibomian glands and Moll and Zeiss gland dysfunction can lead to the development of eyelid abnormalities from the second decade of life.44 Thick cerumen in the ears may lead to obstruction of the external auditory canal and thus hypoacusia.43 Mammary glands may be hypoplastic or even show complete agenesis,45 but sexual development is usually normal.35
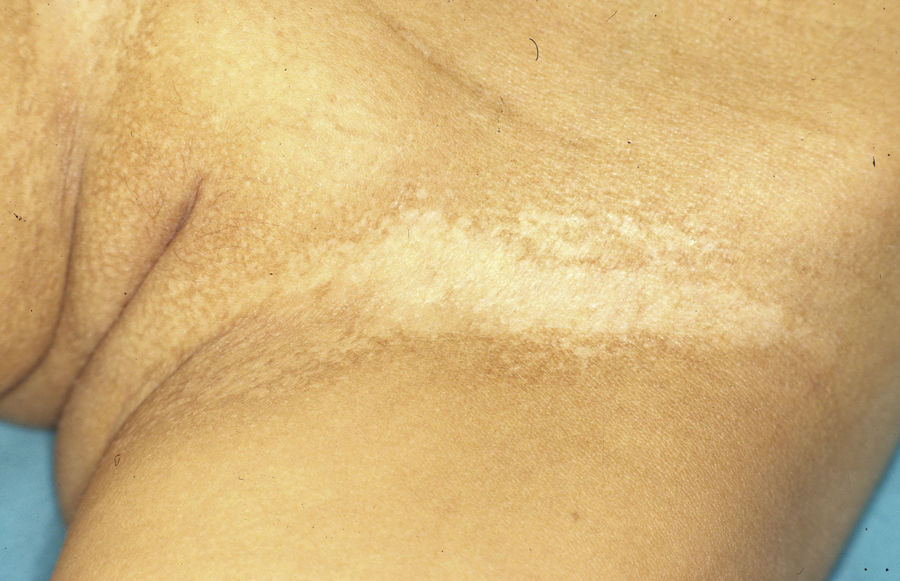
Female carriers of an ED1 mutation may be asymptomatic or show mild or moderate clinical manifestations.46 A characteristic sign suggestive of the disease is present in 70%.47 The most frequent manifestations are tooth abnormalities, mild hypohidrosis, and differing degrees of hypotrichosis.46,48 A patchy distribution of body hair can be found, with somewhat depressed alopecic and xerotic areas in a Blaschkoid distribution alternating with areas of normal skin. The affected areas are more evident with tanning and during childhood.46 In some carriers, a radial shift in the distal phalange of the index finger has been reported.49
HED of autosomal transmissionThe autosomal forms of HED, which are much less frequent than the X-linked forms, can follow a dominant or recessive inheritance, and are caused in most cases by mutations in the EDAR or EDARADD genes.19 Patients with autosomally transmitted HED are clinically indistinguishable from male patients with X-linked HED.50,51 In general, patients with the recessive form have more severe abnormalities, whereas the clinical spectrum of the dominant forms is variable,52,53 and at times very mild, with clinical manifestations similar to those observed in X-linked HED carriers.26,53 In recessive transmission, heterozygous carriers are clinically indistinguishable from individuals with normal genotypes.54 The genetic heterogeneity of HED and the clinical similarity between patients with different transmission modes can be explained by the involvement of ectodysplasin, EDAR, and EDARADD in the same pathway, as activation of NF-κB by ectodysplasin is necessary for the initiation, formation, and differentiation of ectodermal derivatives.54
Diagnosis of HEDEarly diagnosis of HEDs is important to prevent complications essentially resulting from an ineffective control of body temperature during the neonatal period. Diagnosis can be made on clinical grounds in patients with the full syndrome, but it can be difficult in partial cases and in carriers of the disease.34,35 In such cases, it is necessary to perform additional tests to demonstrate decreased sweating or a decreased number of eccrine glands.
The Minor test or iodide-starch test is very useful for confirming the absence of sweating, which is generalized in affected individuals and patchy in female carriers, who have normally functioning eccrine glands alternating with eccrine glands with reduced function in areas with a Blaschkoid distribution (Fig. 7). Examination of large areas of the body, such as the back, can more readily reveal this mosaic distribution. Moreover, such an examination is useful for differentiating between X-linked HED carriers and females affected by autosomally transmitted HED, in whom the function of the sweat glands is almost completely absent.46 Other methods for assessing sweating include iontophoresis after applying pilocarpine to the forearm, sweat pore count, and measurement of skin conductance or temperature; such methods are useful for screening but are less sensitive in patients with residual gland function.40 Skin biopsy is not normally essential for confirmation, but the lack of eccrine glands has a positive predictive value and diagnostic specificity of 100%.37 Molecular analysis is the only way of determining which gene is involved, detecting carriers, and confirming the type of inheritance. This information is vital for genetic counseling.
Prognosis and treatment
For many years, the mortality rate in children with X-linked HED was thought to be around 30% during early childhood,43 but today it is known to be around 13%. Complications appear in the first years of life as a result of hyperthermia and respiratory infections, but after childhood, life expectancy is normal.39 Education of the patients and their families is essential to prevent hyperthermia. Physical activity does not need to be completely avoided, but patients and their families should be aware that high body temperature can cause symptoms such as headache, nausea and vomiting, dizziness, excessive tiredness, and muscle cramps. They should also be made familiar with techniques for reducing body temperature (immersion in water, air conditioning, cold drinks, use of refrigerated devices, etc.). Water sports are ideal for these patients.
There is no treatment for the associated skin disorders or periocular hyperpigmentation, and outbreaks of atopic dermatitis may be difficult to treat. Some authors have suggested these patients have an increased risk of melanoma, and so a full physical examination is recommended once a year.55 Management of children with HED also includes early dental care to prevent maxillary hypoplasia and gum atrophy, which if severe may hinder chewing and language development in addition to being a notable aesthetic problem. Other specialists may also be involved in the care of these patients, for example, ear-nose-throat specialists when nasal and cerumen secretion is a problem, ophthalmologists when eye dryness or problems with the eyelids are present, pulmonologists in the event of respiratory tract infections and, in some case, psychologists.36 Gene therapy with recombinant EDA is still in the experimental phase, but it may offer hope for these patients in the future.56–58 Finally, as is the case in many other genetic diseases, patients diagnosed with HED will need continuous updates about their disease and socioeconomic support. We therefore recommend that patients contact the Spanish Association of Patients With HED (abbreviated as AADE in Spanish): http://www.displasiaectodermica.org.
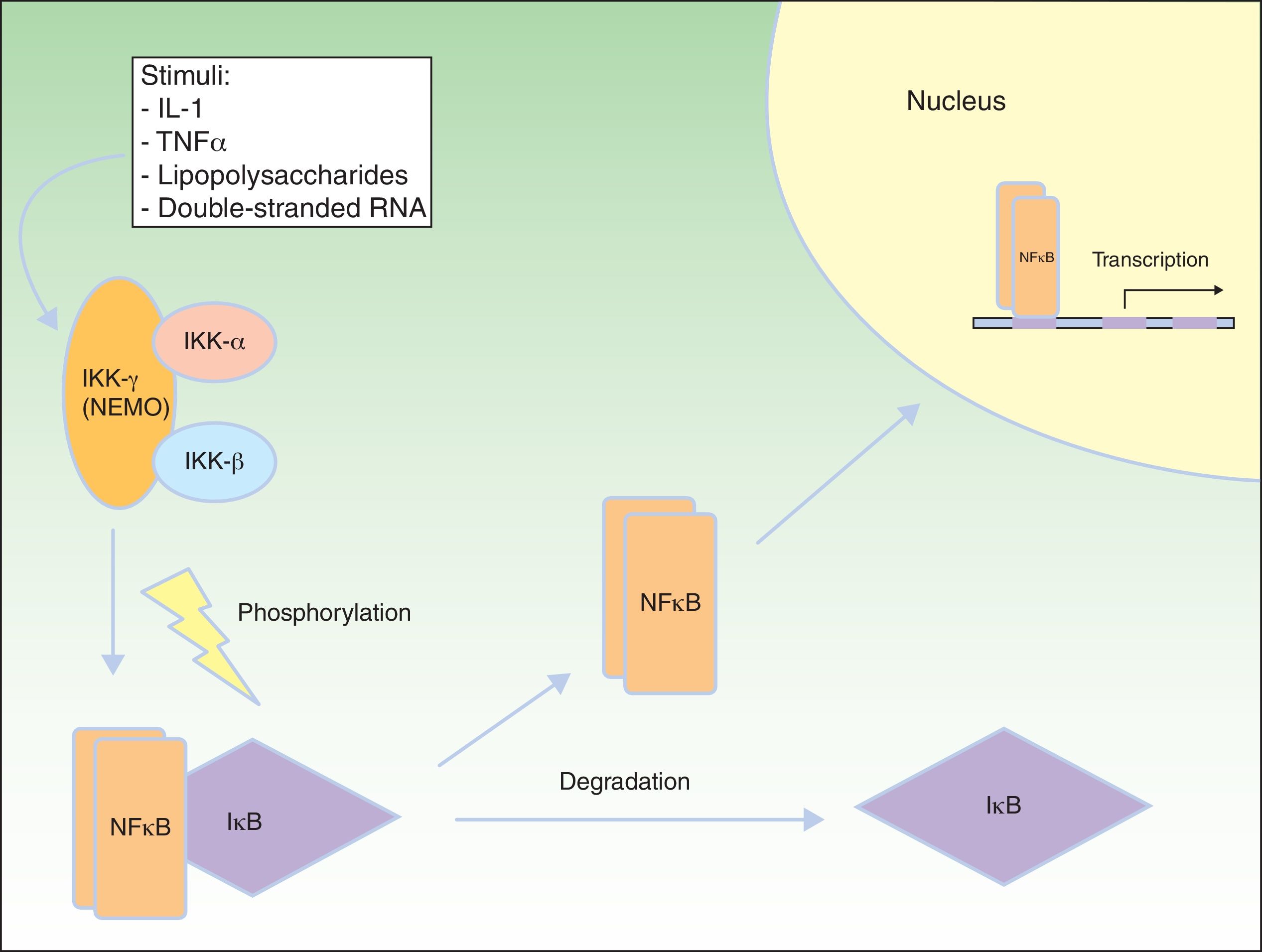
NF-κB Signaling PathwayNF-κB is a transcription factor that regulates the expression of multiple genes implicated in immune and inflammatory responses, reaction to stress, cell adhesion, and protection against apoptosis.59–61 In most cells, NF-κB is kept in an inactive state through cytoplasmic sequestering by the NF-κB inhibitory protein (IkB). Several stimuli, such as interleukin (IL) 1, TNF-α, lipopolysaccharides (bacterial endotoxins), and double-stranded RNA (from viral infections) lead to activation of the cell membrane receptors of the TNF family, such as EDAR and the receptor activator of NF-κB (RANK).62 Activation of these receptors leads to degradation of IkB through the IkB kinase (IkK) complex, which is phosphorylated to form IkB, so allowing NF-κB translocation to the nucleus. In the nucleus, the NF-κB induces genetic transcription, triggering critical inflammatory and immune responses in the development of T and B cells and in osteoclast function and growth of epidermal cells.63 The IkK complex consists of at least 3 subunits: IkK1/IkKα, IkK2/IkKβ, and NEMO/IkKγ (essential modulator of NF-κB). IkK1 and IkK2 act as catalytic subunits, whereas NEMO is a structural and regulatory subunit, essential for the complex to function as a unit. If NEMO is not present, NF-κB shows no response to stimuli,64,65 as NEMO is the main molecule that provides this signaling from the cytoplasm nucleus (Fig. 8).66
![NEMO (nuclear factor [NF] κB essential modulator) and NF-κB pathway. NEMO (also known as NF-κB inhibitory protein [IkB] kinase [IkK] γ) is a regulatory component of the IkK complex. IkB is activated and phosphorylated with different stimuli, leading to its degradation. NF-κB is released and transported to the cell nucleus, where it activates the transcription genes. IL indicates interleukin; TNF, tumor necrosis factor. Adapted from Nelson.73](https://static.elsevier.es/multimedia/15782190/0000010400000006/v2_202210010908/S1578219013001157/v2_202210010908/en/main.assets/gr8.jpeg?xkr=ue/ImdikoIMrsJoerZ+w91sAmkCw32Jed9sZf6jzEuDbFpW7G0NfARZs8afh+9K8v8RN+oFy2ZmalFHXVYo6Ah35yQen1+SXIDS+Cr5GNaVmjb+9Zh6AsajGr7kocMtg1/bDxQAud+COUftU6tKBI1O+aTcsHAN1Oh1IS0yTB+1PUdpKMOhjRc+kc+lwu1SWLp7F65pEmgtBkreo7aBJoxiTwpg/1LI1Xn7zO1BeFXIQoaUErU0nP7j2r/2q2ST+gDfJV0E3uEgLvWFT0pH9tsy31zFbn7nKQAD6UW8Roz0= "NEMO (nuclear factor [NF] κB essential modulator) and NF-κB pathway. NEMO (also known as NF-κB inhibitory protein [IkB] kinase [IkK] γ) is a regulatory component of the IkK complex. IkB is activated and phosphorylated with different stimuli, leading to its degradation. NF-κB is released and transported to the cell nucleus, where it activates the transcription genes. IL indicates interleukin; TNF, tumor necrosis factor. Adapted from Nelson.73")
NEMO (nuclear factor [NF] κB essential modulator) and NF-κB pathway. NEMO (also known as NF-κB inhibitory protein [IkB] kinase [IkK] γ) is a regulatory component of the IkK complex. IkB is activated and phosphorylated with different stimuli, leading to its degradation. NF-κB is released and transported to the cell nucleus, where it activates the transcription genes. IL indicates interleukin; TNF, tumor necrosis factor. Adapted from Nelson.73
Both activation and inhibition of NF-κB have been associated with the development of inflammatory skin conditions.67 Mutations in 2 genes, NEMO and IkBα, have been shown to give rise to a heterogeneous group of genetic disorders that include incontinentia pigmenti; X-linked HED with immune deficiency; osteopetrosis, lymphedema, and HED with immune deficiency; and autosomal dominant HED with immunodeficiency.61,68,69
Incontinentia pigmentiIncontinentia pigmenti is a rare disease whose exact prevalence is not known. It is caused by mutations in the NEMO gene, which is located on the X chromosome (locus Xq28).70,71 Approximately 97% of the patients are female as the condition causes intrauterine death in most males. Most female patients survive due to the selective elimination of cells that express the X chromosome with the mutation.72 The clinical expression of the disease is variable.70,73 Nevertheless, male patients can also have the condition in the case of somatic mosaicism or XXY trisomy.65,74,75
The skin manifestations are the most striking but not the most serious. They are present in almost all patients, with onset during the neonatal period. The lesions are distributed along the Blaschkoid lines and are usually divided into 4 stages: vesicular, verrucous, hyperpigmented, and atrophic (Fig. 9).65 The first stages may go unnoticed, and the patients only show mild hyperpigmented lesions that are not detected until the patient gives birth to an affected daughter. Some women might not even show any clinical manifestations despite being carriers of the mutation.73 The most important clinical problems are vision disorders and neurological deficits, but fortunately, these are less common than the skin manifestations, appearing in 40% and 30% of the patients, respectively.76 In addition, patients may show other problems such as alopecia, tooth abnormalities (cone or peg-shaped teeth, hypodontia, or anodontia), and nail dystrophy.73 Given that a detailed review of this disorder is beyond the scope of this article, we refer the interested reader to other literature sources.
X-Linked HED With Immunodeficiency
X-linked hypohidrotic ectodermal dysplasia with immunodeficiency is a rare X-linked recessive disorder that affects mainly males,77 although some cases have been reported in female patients.78,79 The estimated incidence is 1 case per 250 000 newborns.80 Most patients show small deletions or non-sense mutations in the zinc finger domain (zinc-bound region that allows interaction with other molecules) of NEMO that do not lead to complete loss of NF-κB activation, as occurs with incontinentia pigmenti, but rather an altered or reduced function of the NF-κB pathway.77
Both males and females with X-linked HED with immunodeficiency have mothers with skin lesions reminiscent of incontinentia pigmenti, as well as variable manifestations of HED, including cone-shaped teeth.81,82 Some patients have prominent superficial veins.83 A skin phenotype has also been described with initial involvement of intertriginous areas. The lesions, which have a seborrheic appearance, progress to erythroderma and are reminiscent of lesions in patients with congenital immunodeficiencies such as combined severe immunodeficiency or Omenn syndrome.84 In some cases, incontinentia pigmenti lesions have been observed, thereby illustrating the complexity and overlap between different diseases derived from NEMO mutations.81 Patients with X-linked HED with immunodeficiency have a poor inflammatory response caused by abnormalities in cell response to proinflammatory cytokines (IL-1β, IL-18, and TNF-α).60 The most frequently reported immunodeficiency is dysgammaglobulinemia, with normal or low IgG levels, although elevated IgA, IgM, and IgE levels can also be observed.85 In addition, there have been reports of defects in natural killer (NK) cells, decreased TNF and IL-12 production, abnormal immune response to polysaccharide stimuli with the inability to form specific antibodies to Streptococcus pneumoniae, and a delayed or absent production of isohemaglutinins.77,85,86 Thus, in addition to the characteristic HED phenotype, these patients have serious and recurrent bacterial infections in the lower respiratory tract, skin, soft tissues, bones, gastrointestinal tract, and meninges.65,87 The pathogens are usually pyogenic bacteria such as S pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Klebsiella, Salmonella, and Pseudomonas species, as well as mycobacteria, cytomegalovirus, herpes simplex virus, and Pneumocystis jiroveci.80 Although the infectious complications are generally considered the most life-threatening, defects in NF-κB signaling also increase the risk of inflammatory diseases and inflammatory colitis in particular.80
Female carriers have a range of manifestations, from normal teeth to mild hypodontia or cone-shaped teeth. There has been an anecdotal report of 2 women with mottled skin pigmentation.77
Dominant X-linked HED with immunodeficiencyIn cases of dominant transmission, the patients have an IkBα mutation that prevents phosphorylation and degradation of the IkBα protein resulting in abnormal activation of NF-κB.88 These patients have the classic characteristics of HED, along with T-cell immunodeficiency, giving rise to recurrent infections and immune deficiencies.89 Although patients with NEMO and IkBα mutations share certain clinical manifestations, their immunologic phenotypes are different.90 These patients have marked T-cell deficiency characterized by loss of CD45RO+ cells and abnormal T-cell receptor-mediated stimulation of lymphocytes when studied in vitro. They are unable to respond to TNF-α stimulation, and have abnormal antibody production and greater susceptibility to infections by gram-positive and gram-negative bacteria, as in patients with NEMO mutations. However, these patients have normal NK cell activity, and so are not susceptible to mycobacterial infections.89 It seems that hypomorphic mutations in the stop codon of IkBα could produce a small change in NF-κB activation, giving rise to less severe immunodeficiency.88
Mutations in the stop codon of the NEMO gene yield osteopetrosis, lymphedema, and HED with immunodeficiency.77 As with children with X-linked HED with immunodeficiency, these patients have HED skin manifestations, may have skin lesions similar to those of incontinentia pigmenti, and have an abnormal inflammatory response.85,91 Immunodeficiency is particularly severe, and so these patients experience unusual, aggressive, and often fatal infections from early childhood.60,65 The distinctive clinical characteristics are osteopetrosis and lymphedema, which are thought to be due to abnormal signaling involving RANK, a receptor of the TNF family present in osteoclast progenitor cells.87,92 Differentiation and function of these 2 types of cells depends on NF-κB.69 In these patients, osteoclast differentiation is abolished or severely reduced, and so the bones formed are dense but fragile.92 The NEMO gene encodes the vascular endothelial growth factor receptor 3, an activator of the NF-κB pathway.93 Such a NEMO mutation would be associated with interference in this pathway, giving rise to lymphatic vessel dysfunction and characteristic lymphedema.69
Female carriers of a hypomorphic mutation in the NEMO gene may be asymptomatic or have several manifestations of incontinentia pigmentaria.69,94
Diagnosis of NEMO/IkBα-derived disordersMutations in the NEMO gene should be considered in children with refractory extensive seborrheic or atopic dermatitis, particularly when the facial characteristics of HED are present or the mother has a history of incontinentia pigmenti.84 A biopsy of the skin lesions is recommended in patients with HED with immunodeficiency, as they can develop similar lesions to incontinentia pigmenti after childhood.81
TreatmentThe management of patients with HED manifestations is similar to that presented in the preceding section. Treatment of immunodeficiency may include immune therapies and an aggressive approach to infections, including prophylaxis against gram-positive and gram-negative bacteria, mycobacteria, herpes simplex virus, and Pneumocystis jiroveci.80 Even with such an approach, morbidity and mortality are high.77 Hematopoietic cell transplantation offers the possibility of immune reconstitution, but it brings with it the inherent risks of immunosuppression.69 Little has been published on the subject. Allogeneic transplantation of hematopoietic progenitors could correct the immunodeficiency associated with diseases due to NEMO or IkBα mutation, but the causative mutations do not exclusively affect the hematopoietic system, and so constitutional manifestations not related to immunodeficiency are not corrected by transplantation. Inflammatory colitis might therefore even worsen on correcting immunodeficiency.95
Changes in the Transcription and/or Expression of Regulators of Certain GenesDisorders Derived From p63 MutationsThe p63 gene, also known as TP63 (tumor protein 63), is located on chromosome 3 (locus 3q27) and encodes the transcription factor p63, involved in ectodermal development. The regions of greatest biological importance are the DNA binding domain (a region that allows transactivation through binding of p63 to DNA), the sterile alpha motif (SAM), which is thought to participate in protein-protein interactions, and the transactivation inhibition domain (TID), which is located next to the SAM domain and could be involved in balancing the effects of different isoforms of TP63.96 The p63 protein is expressed very early during embryogenesis and plays an essential role in inducing epidermal differentiation and proliferation and in other processes including facial and limb development.96–98 Lack of expression of this protein during early development of ectodermal structures might block a chain of interactions between the epithelium and the mesenchyme, thereby interfering in normal morphogenesis.99 In addition, p63 regulates expression of P-cadherin, a critical regulator of hair development.100
Heterozygous mutations in the p63 gene are responsible for at least 6 different syndromes that combine ED, orofacial clefts, and limb malformations.101 The ectrodactyly-ectodermal dysplasia-cleft lip/palate syndrome (EEC) syndrome is the prototype syndrome. Other syndromes include ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC) syndrome, limb-mammary syndrome, acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome, Rapp-Hodgkin syndrome, and split-hand/foot malformation. All these syndromes present at least 1 of the key characteristics of EEC syndrome.101 There is a strong genotype-phenotype correlation that is dependent on the location of the p63 mutation.96 The most frequently mutated amino acid residues are R204, R227, R279, R280, and R304. The phenotype related to the R204 mutation is very similar to the complete phenotype of EEC syndrome, but the patients have a lower frequency of hypohidrosis and orofacial cleft; the R227 mutations are rarely associated with orofacial cleft or syndactyly, but the incidence of renal problems and hypohidrosis is higher whereas hearing disorders are absent; the R279 mutation is the only one that gives rise to ankyloblepharon, which is usually associated with ectrodactyly (deformity of the limbs in which part or all of the fingers are missing, giving the hands or feet the form of a lobster claw); patients with the R280 mutation frequently have skin manifestations and syndactyly but hypohidrosis and hearing or renal disorders are absent; and finally, the R304 mutation is associated with a higher percentage of orofacial cleft, syndactyly, and hearing disorders.101
Ectrodactyly-Ectodermal Dysplasia-Cleft Lip/Palate SyndromeEEC syndrome is relatively common. Although it can appear in sporadic cases, autosomal dominant inheritance can be detected in most cases.102 Mutations in the p63 gene are present in 98% of patients with the classic phenotype; these mutations are generally point mutations in the DNA binding domain, while mutations in the SAM and TID domains are rare.96 At least 30 different mutations have been identified, of which 5 (mutations in amino acids R204, R227, R279, R280, and R304 of p63) are responsible for 86% of the cases.101
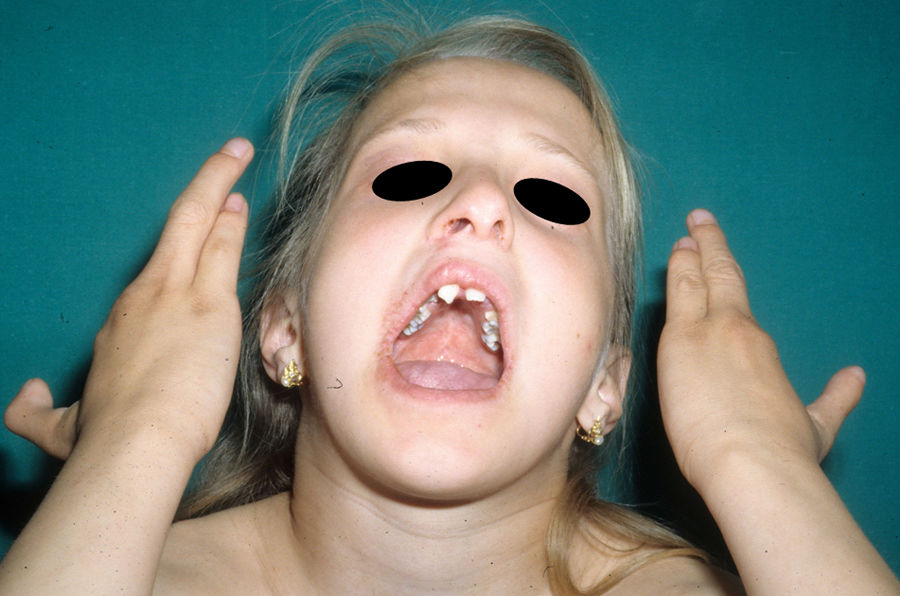
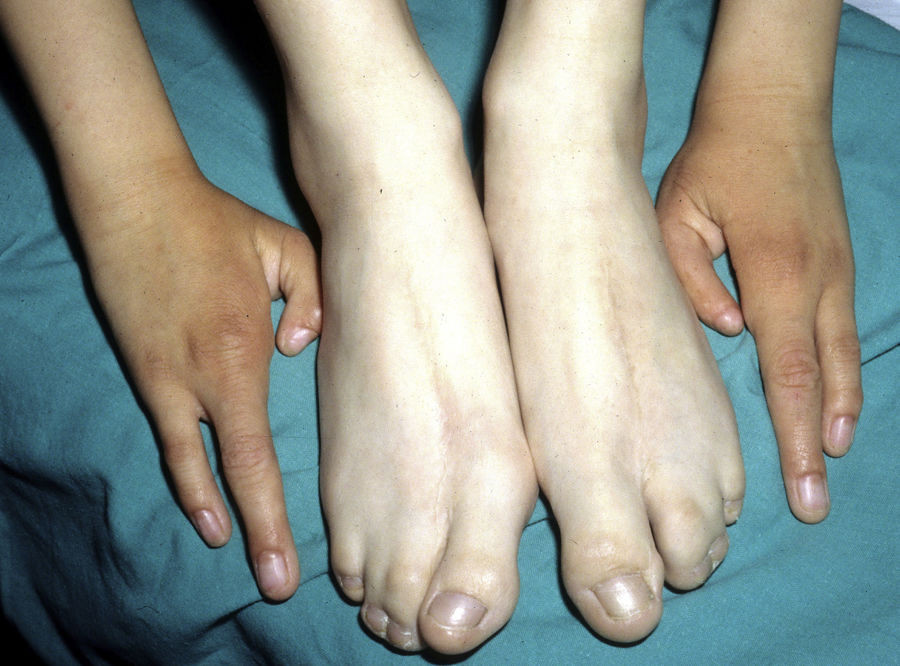
The most frequent abnormalities are malformations of the limbs, ED, and orofacial cleft (Fig. 10), followed by tear duct abnormalities, genital malformations, and deafness, although the clinical manifestations vary greatly within and among families.103 Among the most representative limb malformations are ectrodactyly and syndactyly (Fig. 11). Ectodermal dysplasia may be associated with sparse, hypopigmented hair, absence of eyebrows and eyelashes, and alopecia. The skin is usually fine, dry, and of an atopic appearance, while the nails are dystrophic and may appear pitted. Perioral lesions and angular cheilitis may be present in the oral commissures as a result of anatomic changes caused by reconstructive surgery for cleft lip/palate.104,105 Tooth abnormalities such as hypodontia or anodontia have also be reported, as well as a propensity to tooth decay due to defective enamel and changes in the salivary gland function. Orofacial cleft is common and may be accompanied by maxillary and malar hypoplasia. Tear duct stenosis contributes to keratitis, which is sometimes associated with photophobia.101 Urogenital and anogenital abnormalities (micropenis, hypospadia, vaginal septum, and female genital hypoplasia), hypothalamic-hypophyseal insufficiency, thymic hypoplasia, and mental retardation may also be present.106,107 Isolated cases have been reported of an association with white sponge nevus,108 as well as micrognathia, cleft soft palate, and glosptosis (Pierre Robin sequence), but the relevance of this association is not known.109


Prenatal ultrasound diagnosis is an important aspect of diagnosis of EEC syndrome, not only because ectrodactyly and cleft lip/palate may alert the physician, but also because severe associated genitourinary abnormalities can be detected.107
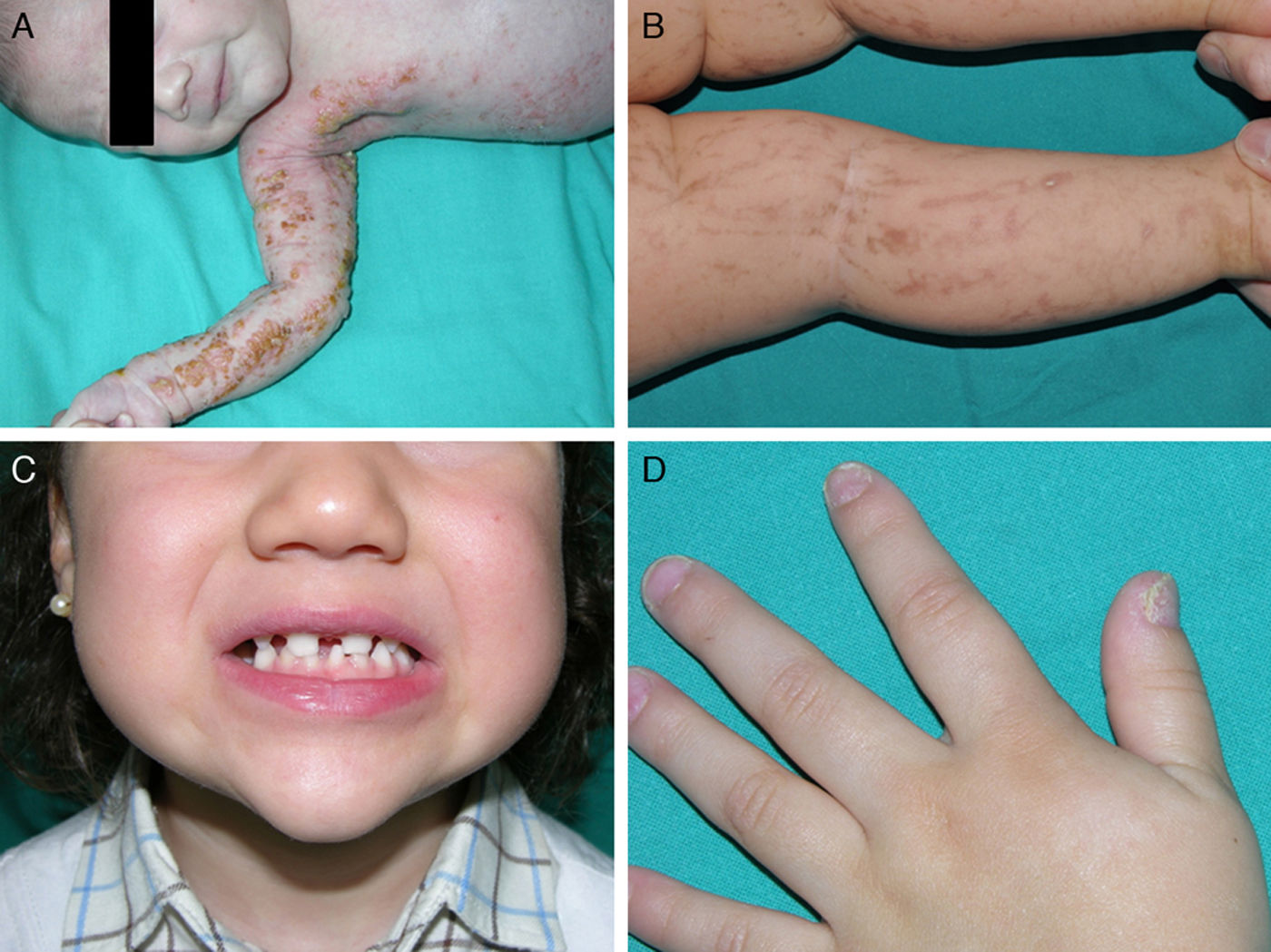
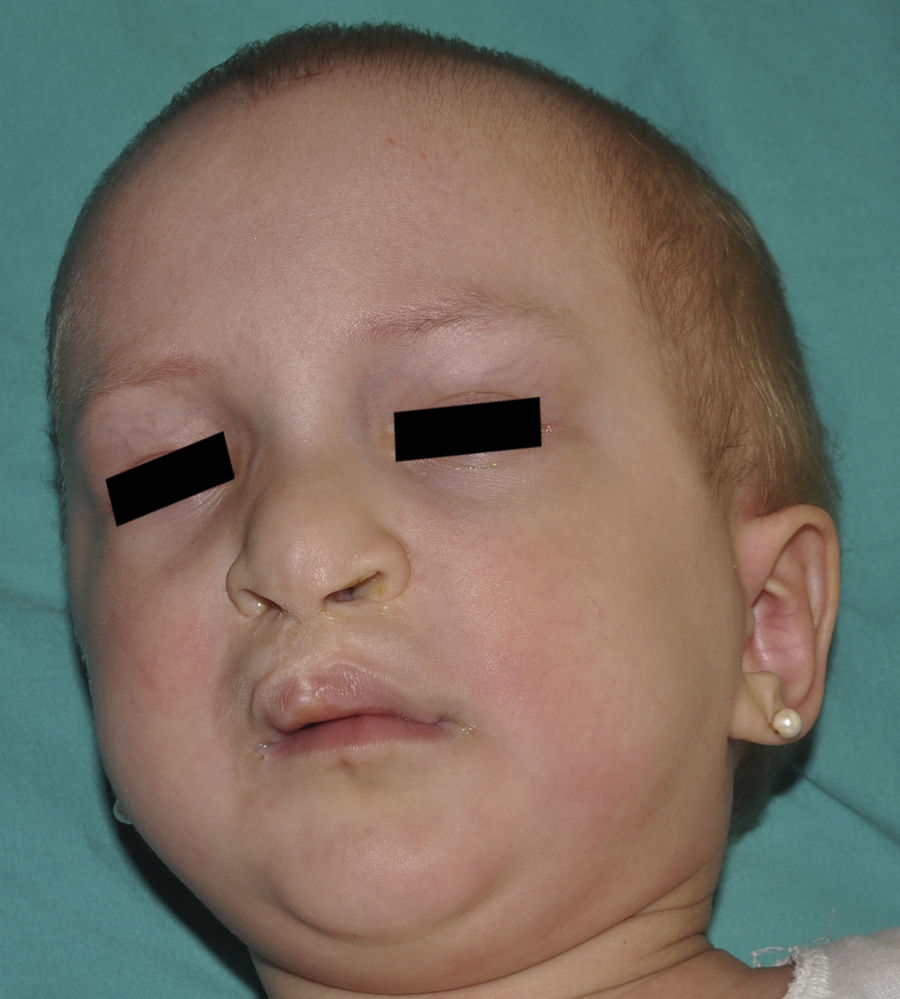
Ankyloblepharon-ectodermal dysplasia-cleft lip/palate syndromeThe AEC disorder follows an autosomal dominant transmission pattern and results from non-sense mutations in the SAM domain of protein p63, implicated in the interaction with other proteins participating in the regulation of transcription and development of skin appendages. The characteristic clinical triad consists of ankyloblepharon, ectodermal defects, and cleft lip/palate (Fig. 12).110 Ankyloblepharon is a condition in which fibrous bands between the eyelids prevent these from opening or moving normally. The ectodermal abnormalities are similar to those of other EDs.96 In addition to these abnormalities, skin erosions can be observed in the scalp, head, neck, skin folds, palms, and soles. The erosions heal poorly and tend to become superinfected. On the upper part of the trunk, they heal to leave residual scarring of a cribriform, reticular, stellate, or punctate pattern. Although the exact reasons for the erosions and delayed wound healing are not well known, a contributing factor could be the role of protein p63 in the formation of basal cells and epidermal differentiation.111 At birth, patients may present manifestations such as erosive lesions, congenital erythroderma, ichthyosiform scaling, and even collodion membrane which may lead to an initial suspicion of epidermolysis bullosa or a keratinization disorder.111 Changes in pigmentation (hyperpigmentation or hypopigmentation) are also frequent (Fig. 13). Reticular hyperpigmentation, which becomes more accentuated with age, can be observed in the large skin folds, whereas characteristic hypopigmentation, which improves as the patient gets older, may be present around the eyes giving a mask-like appearance. Almost all patients have sweating disorders.111 The extent of alopecia is variable and does not seem to be related to age or severity of prior erosions in the scalp. Hair may be thick, wiry, fragile, or matt, and have variable or even 2-tone pigmentation (pigmented and pale hairs). Hair shaft abnormalities, such as pili annulati, pili torti, and pili canaliculi, and irregular indentation may be present.112 Nail involvement is also variable (Fig. 14) and ranges from complete anonychia to mild scaling of the nail plate.111 Other skin abnormalities occasionally reported include absence of dermatoglyphs, palmoplantar hyperkeratosis, punctate keratoderma, hyperlinearity, and hyperkeratosis on the elbows and knees.111
 cleft lip in a patient with ankyloblepharon-ectodermal dysplasia-cleft lip/palate syndrome.")


Facial dysmorphia includes, in addition to cleft palate and lips, small or malformed ears, maxillary hypoplasia, lack of permanent teeth, and decreased length of the eyelid skin crease. Failure to thrive, syndactyly, nasolacrimal atresia, recurrent otitis media, hearing loss, and hypospadias are also common.96 Some individuals may experience neutropenia of unknown origin and have recurrent skin and ear infections, which may progress to bacteriemia and sepsis.96
AEC syndrome should be suspected in any neonate with erythroderma and cleft lip/palate, particularly when other skin manifestations are present, such as erosions in the scalp or skin, although ankyloblepharon is not always present. Skin biopsy, although not very specific, shows mild hyperkeratosis, epidermal atrophy, basal pigmentation, and/or incontinentia pigmenti, and a prominent superficial vascular plexus with mild perivascular infiltrate consisting mainly of lymphocytes.112
The first objective of treatment is to prevent skin erosions, and so energetic cleansing of the skin should be avoided. Secondary wound infection can be hard to treat, and so extensive daily hygiene and application of antiseptics to the erosions are recommended. The administration of antibiotics for long periods would only seem to provide a minimal or temporal improvement in the lesions. Some authors have suggested that the use of corticosteroids113,114 or low doses of doxicycline115 may be useful for reducing inflammation and improving healing, but the risk of side effects should be taken into consideration. Looking to the future, other therapeutic strategies, such as gene therapy and use of epidermal stem cells to regenerate affected skin, are under investigation.111
Acro-dermato-ungual-lacrimal-tooth syndromeEEC and ADULT syndromes are considered allelic disorders with overlapping clinical characteristics, such as ectodermal, limb, and tooth abnormalities. The main difference lies in the absence of orofacial cleft from ADULT syndrome.116 ADULT syndrome arises through mutations that affect the TID domain, implicated in transactivation of the p63 gene.117 To date, 5 mutations in this gene have been reported; 2 of these have also been identified in patients with EEC syndrome.118
Limb-mammary syndromeThe limb-mammary syndrome, with an autosomal dominant inheritance, is caused by mutations in the SAM and TID domains of the p63 gene.119 Its clinical characteristics overlap with those of EEC, AEC, ADULT and Rapp-Hodgkin syndromes, as well as with ulnar-mammary syndrome (caused by mutations in the TBX3 gene at locus 12q24.1).120 The syndrome is characterized by ectrodactyly, hypoplasia of the mammary glands and nipples, cleft palate (without a cleft lip), and the absence of skin or hair abnormalities. Other manifestations include lacrimal duct stenosis, hearing loss, urogenital abnormalities, nose dysplasia, hypohidrosis, hypodontia, and gonadal dysplasia.99,120
Differentiation between limb-mammary syndrome and EEC syndrome is based on 3 findings120: hypoplasia of the mammary glands and nipples, present in all cases of limb-mammary syndrome but only occasionally in EEC syndrome; hair and skin abnormalities, absent in patients with limb-mammary syndrome; and cleft lip, present only in patients with EEC syndrome.
Rapp-Hodgkin syndromeRapp-Hodgkin syndrome is a disorder of autosomal dominant transmission,121 produced by mutations that, like in AEC syndrome, affect the SAM domain, thereby explaining the considerable clinical and molecular overlap between the 2 syndromes.96,122 Some authors consider these 2 syndromes as the same disorder.121–127
Tricho-dento-osseus syndromeThe tricho-dento-osseus syndrome is a condition of autosomal dominant transmission caused by different mutations in the DLX3 gene, located on chromosome 17 (locus 17q21). This gene, which is also responsible for imperfect amelogenesis, encodes the DLX3 protein,128 which is expressed during embryogenesis and which participates in the differentiation of tissue derived from the ectoderm, bone tissue, and cartilaginous tissue.129 The clinical manifestations are variable and include abnormalities in tooth enamel, nail abnormalities, blond curly hair, sclerosis and thickening of cranial bones, radiographic abnormalities such as hypocalcification, and taurodontism.130
Witkop SyndromeWitkop syndrome, an autosomal dominant condition also known as the tooth and nail syndrome and nail dysgenesis and hypodontia, is caused by a mutation in the MSX1 gene, located on chromosome 4 (locus 4p16.1). The MSX1 gene participates in the formation of certain teeth (premolars, first molars, and third molars) and nails, by determining the thickness and integrity of the nail plate.131 Typical clinical characteristics are nail dysplasia and hypodontia, although the clinical manifestations are very varied. In some cases, hair abnormalities (fine or thick hair) have been reported, though most individuals have normal hair and sweat gland function.132
Ellis-van Creveld SyndromeEllis-van Creveld syndrome follows an autosomal recessive transmission pattern and is caused by mutations in the EVC or EVC2 genes; the clinical phenotype is the same for both types of mutation.133–135Approximately 30% of patients with Ellis-van Creveld syndrome have no mutations in either of these genes, suggesting that greater genetic heterogeneity may be present.136 The syndrome is characterized by bone abnormalities, nail dysplasia, orofacial abnormalities, and cardiovascular malformations.137 Patients have a short stature, acromesomelic limb shortening (more prominent in the distal region), and a narrow chest. Polydactyly, syndactyly, genus valgum, and various types of tooth abnormalities are common.137,138
Weyers Acrodental DysostosisWeyers acrodental dysostosis is an autosomal dominant disorder whose causative mutations are located in the EVC and EVC2 genes.133,134,137 The transmission pattern and milder phenotype distinguish this condition from Ellis-van Creveld syndrome.133 Clinical expression is variable and characterized by short stature, hypotelorism, prominent ears, postaxial polydactyly, oral abnormalities (irregular, small, peg-shaped teeth and hypodontia), and onychodystrophy (dysplastic or hypoplastic nails).133,137
Group 2Hidrotic Ectodermal DysplasiaHidrotic ectodermal dysplasia, also known as Clouston syndrome, is caused by mutations in the GJB6 gene which is located on chromosome 13 (locus 13q11-q12.1) and which encodes connexin 30.139 This autosomal dominant disorder is particularly common among French-Canadian individuals,140 and only appears very exceptionally de novo.141 Mutations in this gene can also give rise to other disorders such as autosomal dominant and autosomal recessive sensorineural deafness.142
Connexins are transmembrane proteins that facilitate intercellular communication.140 Connexin molecules form hexamers called connexons, which interact with other connexons of adjacent cells to form intercellular gap junctions.142 These channels allow the diffusion of small molecules between cells and mediate signal and nutrient exchange, thereby coordinating cell activities and response to stimuli. More than 10 different connexins have been identified in skin. Each connexin seems to have its own specific properties, and mutations are responsible for a distinct skin disorder. Thus, hidrotic ectodermal dysplasia and KID syndrome140 are caused by mutations in the GJB6 and GBJ2 genes, which encode connexin 30 and connexin 26, respectively.142 These 2 connexins share 76% of their amino acid sequence and are coexpressed in the stratum corneum, sweat glands, and hair follicles.140 This overlap, despite the different tissue expression of connexin 26 and connexin 30, implies that certain functions are shared and that there is perhaps a direct interaction between the 2 proteins in many ectodermal epithelia. In fact, both are associated with certain clinical characteristics such as nail dystrophy, hair loss, and plantar keratoderma, and recently there has been a report of a patient with a connexin 30 mutation whose clinical manifestations were similar to those of keratitis-ichthyosis-deafness (KID) syndrome.140
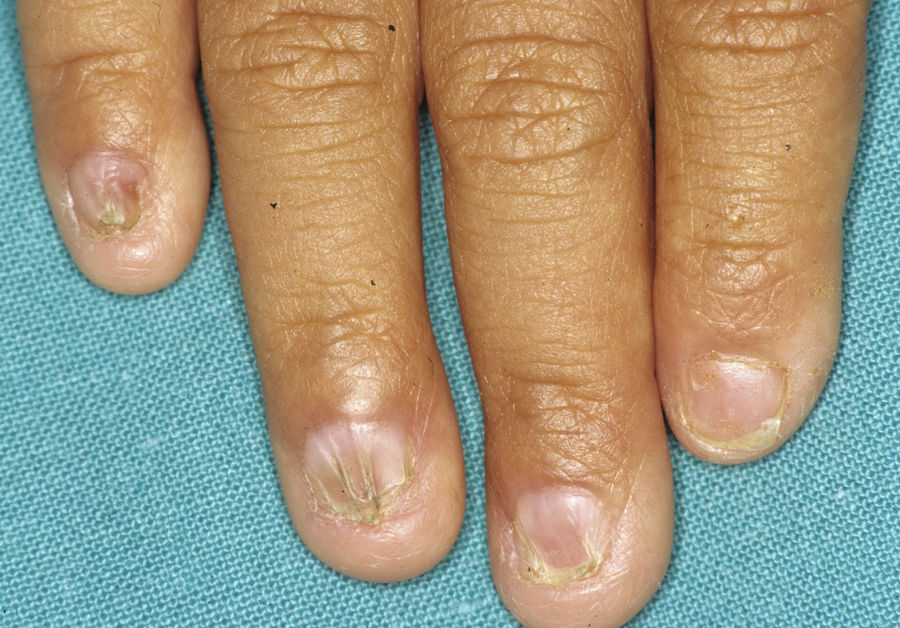
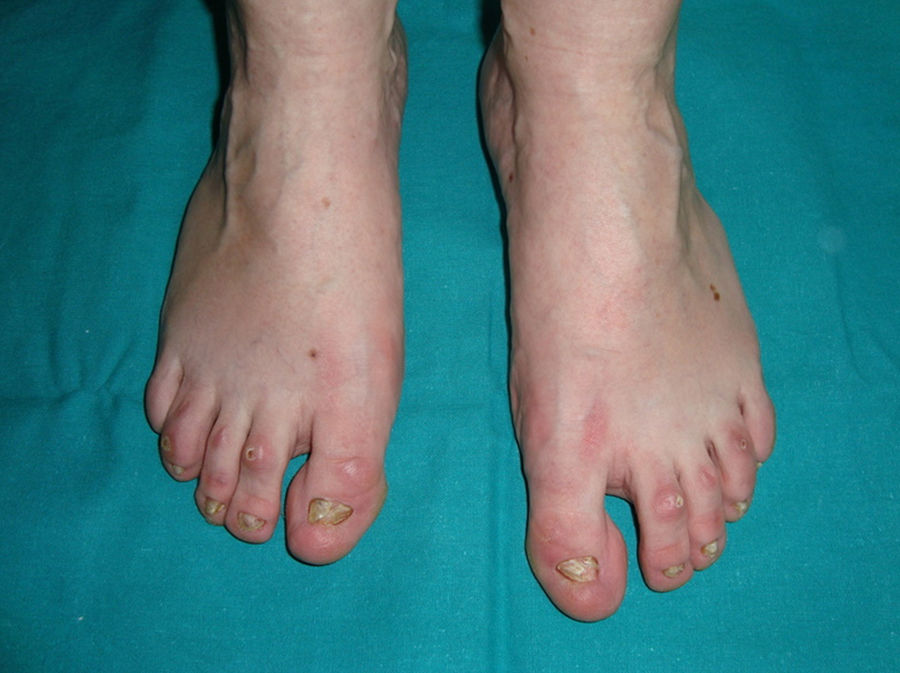
The 3 main clinical characteristics of hidrotic ectodermal dysplasia are hair loss, nail dystrophy, and palmoplantar keratoderma (Figs. 15 and 16).140 Unlike patients with HED, patients with hidrotic ectodermal dysplasia have normal teeth and sweat and sebaceous gland function.140,143 Hair abnormalities are manifest as atrichia or hypotrichosis, which may progressively worsen140; the fine, slow-growing hair has a disorganized structure and reduced birefrigence.144 Women are completely bald, whereas men show expression that varies from fair hair with focal alopecia to complete baldness.145 The eyebrows and eyelashes are scant or absent, as is pubic and axillary hair.140 Nail disorders range from an almost normal appearance to micronychia or anonychia; the nail plate may show thickening, desquamation, color changes, striation, and onycholysis.140 Nail abnormalities in these patients may be reminiscent of congenital pachyonychia.146 Some patients also present diffuse palmoplantar keratoderma and discrete skin hyperpigmentation, which is particularly evident underneath the free edge of the nails and on the finger and toe joints, knees, and elbows.140,147 Eccrine syringofibroadenomas have been reported in several patients,148,149 and there has also been an isolated case of congenital pseudoainhum.150 These clinical manifestations may be accompanied by strabism, conjunctivitis, pterygium, cataracts,143 sensorineural deafness, polydactyly, and syndactyly,147 but facial dysmorphism is not present and general physical development is normal.
.")
.")
The autosomal recessive disorder Zlotogora-Ogur syndrome is caused by mutations in the poliovirus receptor-like 1 (PVRL1) gene, located on chromosome 11 (locus 11q23.2), which encodes the nectin-1 protein.151 Nectins are calcium-independent cell-cell adhesion molecules that act at cell-cell junctions sometimes in conjunction with cadherins. So far, 4 different types of nectins have been reported.152 Nectin-1 is expressed in several ectodermal tissues, including the skin, teeth, and hair, essentially within the stratum spinosum.153 Mutation in this gene also gives rise to nonsyndromic orofacial cleft type 7.154 Characteristic clinical findings include manifestations of ectodermal dysplasia, bilateral cleft lip/palate, mental retardation, and syndactyly.155 Hair is sparse and short, and when individuals are over 40 years, they may become completely bald. Eyebrows are also sparse, especially in the lateral regions. Xerosis, hypoplastic dermatoglyphs, and progressive palmoplantar hyperkeratosis are also observed. Tooth abnormalities include delay in dental eruption, microdontia, hypodontia, and anodontia. Nails are normal or mildly dysplastic. Patients have peculiar facial features, with an oval face and large anteverted ears. Syndactyly, which may be partial, is often present in the second, third, and fourth fingers, and can sometimes affect both hands.151 Other manifestations include deafness, genitourinary or renal abnormalities, nipple abnormalities and lumbar lordosis, and variable mental retardation.156 Differential diagnosis should be established mainly with EEC syndrome, with which it shares most manifestations. The main difference is in the transmission pattern (EEC syndrome follows autosomal dominant inheritance) and limb malformations, mainly ectrodactyly, which are present in 85% of the patients with EEC syndrome.103,156
In August 2010, a second nectin-derived condition was reported arising from an abnormality in nectin 4, which is encoded by PVRL4.152 This syndrome is similar to the one described above, but there is no cleft palate and it is known as ectodermal dysplasia-syndactyly syndrome.
Ectodermal Dysplasia-Fragile Skin SyndromeEctodermal dysplasia-fragile skin syndrome is an autosomal recessive condition that was recently reclassified as a epidermolysis bullosa simplex. It is caused by mutation in the plakophilin gene (PKP1), located on chromosome 1 (locus 1q32).157 PKP1 is a structural component of desmosomes and is expressed in the stratified squamous epithelium, the myocardium, the meninges, and part of the lymph nodes.158 Like other types of epidermolysis bullosa, the condition is characterized by substantial trauma-induced skin fragility, generalized erythema, alopecia, nail dystrophy, and focal keratoderma with painful fissures (Fig. 17). Some patients present hypohidrosis, but the teeth are normal in all cases.157 Histologic study shows widened intercellular spaces, separation of keratinocytes, intraepidermal clefts, acantholytic keratinocytes, and different degrees of dyskeratosis,159 but to date the development of skin carcinomas has not been reported.
Ectodermal Dysplasia-Ectrodactyly-Macular Dystrophy Syndrome
Ectodermal dysplasia-ectrodactyly-macular dystrophy (EEM) syndrome is an autosomal recessive disorder caused by a mutation in the CDH3 gene, located on chromosome 16 (locus 16q22.1); this gene encodes the cadherin-3 protein.160,161 Cadherins are calcium-dependent adhesion molecules with several extracellular domains, a transmembrane region, and an intracellular region. The intracellular region binds to β-catenin, which is implicated in transcription and cellular adhesion. Expression of CDH3 during embryogenesis is important for normal development. It is expressed at least in the orofacial region and pharyngeal arches, as well as in the limbs, and it may play an important role in the morphology of the human hand.161 EEM syndrome is characterized by syndactyly, retinal degeneration, and sparse hair,160 although the clinical manifestations are variable. In addition to syndactyly, which may be bilateral, 1 or several phalanges may be missing or fingers or toes may be completely hypoplastic. The hands are usually more severely affected than the feet.161 Another key sign is progressive retinal degeneration, with gradual vision loss. Prominent pigmentation is observed in the posterior pole of the retina, along with macular atrophy. Patients may also have hypotrichosis with sparsely populated eyebrows, few eyelashes, and tooth abnormalities (hypodontia and small and very separated teeth).162
Odonto-Onycho-Dermal DysplasiaThe autosomal recessive disorder, odonto-onycho-dermal dysplasia, is due to mutations in the WNT10A gene, located on chromosome 2 (locus 2q35).163 The WNT genes encode a large family of glycoproteins implicated in a signaling pathway crucial for the development of ectodermal-derived tissue during embryogenesis and adult life.165,165 Thus, WNT10A is important for tooth and hair follicle formation, and for epidermal regeneration, lingual papillae formation, and sweat gland function.164 Although the condition is termed odonto-onycho-dermal dysplasia, tricho-onyco-dermal dysplasia would be more appropriate given the hair involvement.163 Characteristic skin lesions include the appearance in the facial region of reticular, erythematous, telangiectatic, atrophic plaques, which become more intense with heat.166,167 Another frequent observation is palmoplantar hyperkeratosis, accompanied by hyperhidrosis and painful fissuring.168,169 Other abnormalities include the presence of smooth tongue with reduced or absent papillae, pilaris keratosis,170 hypotrichosis, and tooth and nail abnormalities.167 Some patients may be slightly mentally retarded.169 Different mutations in this same gene are responsible for another autosomal recessive ectodermal dysplasia, Schopf-Schulz-Passarge syndrome. This syndrome shares certain clinical features (hypodontia, nail dystrophy, palmoplantar keratoderma, smooth tongue, hyperhidrosis, and hypotrichosis), but in addition, patients have other features such as cysts in the eyelids and an increased risk of developing skin tumors.165,171
In conclusion, ectodermal dysplasias are a heterogeneous group of hereditary disorders that bear many similarities and are difficult to classify. Biomolecular findings in recent years have brought us closer to a useful clinical-functional classification in clinical practice given that different genetic abnormalities in different functional pathways can be linked to a given phenotype. However, many disorders within the group of ectodermal dysplasias have yet to be studied or identified. Nevertheless, the dermatologist should be aware of the main signs and symptoms of these disorders when searching for a diagnosis. In addition, carriers should be identified so that genetic counseling can be offered.
Ethical responsibilitiesProtection of human and animal subjects.The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of data.The authors declare that they have followed their hospital's protocol on the publication of data concerning patients and that all patients included in the study have received sufficient information and have given their written informed consent to participate in the study.
Right to privacy and informed consent.The authors obtained the informed consent of patients and/or subjects mentioned in this article. The informed consent form is located in the archives of the corresponding author.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: García-Martín P, et al. Displasias ectodérmicas: revisión clínica y molecular. Actas Dermosifiliogr. 2013;104:451–70.



