





Vasculitis is a term that refers to damage and inflammation of the walls of blood vessels of any size. The classification of types of cutaneous vasculitis continues to be a challenge, probably because of our lack of understanding of the etiology and pathogenesis of this condition. Changes in the vessel wall will be visible on microscopy and will enable the different clinical forms to be distinguished according to the caliber of affected vessels, the type of cell that predominates in the inflammatory infiltrate, or the presence of such key findings as extravascular granulomas. Skin manifestations (macules, papules, nodules, livedo reticularis, etc.) correlate with the size of the vessel affected. The prognosis in cases of vasculitis with skin involvement will be determined by the presence or absence of extracutaneous disease. Systemic vasculitis shows a predilection for certain organs, such as the kidney or lung. The introduction of immunosuppressant drug treatments has led to evident improvement in survival rates for patients with vasculitis. This review covers practical aspects of the pathophysiology, histopathology, treatment, and differential diagnosis of the main clinical presentations of vasculitis with cutaneous involvement.
El término vasculitis define el daño e inflamación de las paredes vasculares, con independencia del calibre del vaso afectado. Clasificar las vasculitis con afectación cutánea constituye un reto aún no superado, como consecuencia, probablemente, de la falta de conocimiento sobre su etiopatogenia. En la microscopía deben encontrarse datos de afectación de la pared vascular, diferenciándose unos cuadros de otros por el calibre del vaso afectado, el tipo celular predominante en el infiltrado inflamatorio o la presencia de hallazgos característicos, como los granulomas extravasculares. En la clínica existe una correlación entre los hallazgos cutáneos (máculas, pápulas, nódulos, livedo reticular…) y el calibre del vaso afectado. El pronóstico de las vasculitis con afectación cutánea viene determinado por la presencia o no de compromiso extracutáneo, de forma que las vasculitis sistémicas muestran especial predilección por órganos como el riñón o el pulmón. No obstante, tras la introducción de los fármacos inmunosupresores la supervivencia de estos pacientes ha experimentado un cambio ostensible. En este artículo se revisan de forma práctica los aspectos relacionados con la fisiopatología, histopatología, tratamiento y diagnóstico diferencial de los principales cuadros vasculíticos con afectación cutánea.
Vasculitis is a term that refers to the inflammation and necrosis of blood vessels irrespective of the type of vessel involved (veins, arteries, or both), the etiology of the process, or the organ affected. The condition can be idiopathic or secondary to infection, drugs, neoplastic disease, or systemic inflammatory disease.
Cutaneous involvement is very common in the different types of vasculitis. Skin lesions may be the only manifestation of the disorder or they may occur in the context of systemic disease, but there are no clinical, histologic, or laboratory criteria that differentiate with certainty between vasculitides of the skin and systemic vasculitides.
ClassificationThe development of a standard classification scheme that would be acceptable to all the professionals involved in the diagnosis and management of the different types of vasculitis continues to present a challenge.1,2
The first diagnostic classification, which was drawn up by Zeek et al.3 in 1950, was based on the size of the vessel involved and has served as a basis for later schemes.4
In 1990, the American College of Rheumatology published a classification scheme based on a set of clinical and histopathologic criteria and first used the term hypersensitivity vasculitis to designate small vessel cutaneous vasculitis.5 In 1994, the Chapel Hill Consensus Conference decided to abandon clinical criteria and standardize the nomenclature on the basis of histopathologic characteristics, grouping the different entities according to the caliber of the affected vessel.6 However, none of these systems have managed to overcome the difficulties posed by the overlapping clinical features of different entities or to deal adequately with the question of the immunopathologic mechanisms involved and their correlation with prognosis.7 In an effort to overcome these shortcomings, Carlson et al.8 proposed a framework for classifying cutaneous vasculitides that includes clinical features, histopathologic criteria, laboratory findings, and the relationship of all of these aspects with the different etiologies (Table 1). Unfortunately, in spite of all this work, there is still no universally accepted classification scheme for vasculitides.
Classification of Vasculitides.
| 1. Small vessel vasculitis |
| 1.1. Neutrophilic vasculitides (mediated by immune complexes) |
| A) Cutaneous leukocytoclastic vasculitis or hypersensitivity vasculitis |
| B) Henoch–Schönlein purpura |
| C) Urticarial vasculitis |
| D) Erythema elevatum diutinum |
| E) Acute hemorrhagic edema of infancy |
| 1.2. Eosinophilic vasculitides |
| A) Hypereosinophilic syndrome |
| B) Vasculitis associated with connective tissue disease |
| 1.3. Lymphocytic vasculitides |
| A) Pityriasis lichenoides et varioliformis acuta |
| B) Perniosis |
| 2. Small to medium-sized vessels |
| 2.1. Neutrophilic vasculitides |
| 2.1.1. Mediated by immune complexes |
| A) Cryoglobulinemic vasculitis |
| B) Vasculitis associated with connective tissue disease |
| C) Hypocomplementemic form of urticarial vasculitis |
| D) Septic vasculitis |
| 2.1.2. Mediated by antineutrophil cytoplasmic antibodies (ANCA) |
| A) Microscopic polyangiitis |
| B) Wegener granulomatosis |
| C) Churg–Strauss purpura |
| D) Drug-induced ANCA-positive vasculitis |
| 2.2. Lymphocytic vasculitides |
| A) Degos disease |
| B) Viral and rickettsial infections |
| C) Vasculitis associated with connective tissue disease |
| 3. Medium to large vessels |
| 3.1. Neutrophilic vasculitides |
| A) Polyarteritis nodosa (classic and cutaneous) |
| B) Nodular vasculitis or erythema induratum of Bazin |
| 3.2. Granulomatous vasculitides |
| A) Giant cell arteritis |
| B) Takayasu arteritis |
| 3.3. Lymphocytic vasculitides |
| Kawasaki disease |
Adapted from Carlson et al.8
The development of the different types of vasculitis is affected by many factors. One of these is the deposition of circulating immune complexes within vessel walls, a mechanism implicated in hypersensitivity vasculitis. Potential antigens include drugs and chemicals as well as infectious agents such as viruses or bacteria. Interacting with the complement system, the immune complex deposition stimulates the production of chemotactic factors, vasoactive amines (histamine), and proinflammatory cytokines (interleukin [IL] 1, tumor necrosis factor α), which in turn induce the expression of adhesion molecules on endothelial cells (intracellular adhesion molecule-1, vascular cell adhesion molecule-1, P-selectin, and E-selectin). The hypothesis is that this phenomenon promotes the recruitment of neutrophils, which subsequently degranulate by binding with the Fc portion of the deposited antibodies and release reactive oxygen species, collagenase and elastase, which trigger fibrinoid necrosis of the vessel walls.9
Antineutrophil cytoplasmic antibodies (ANCA) also play a role in the development of vasculitis. ANCA are autoantibodies directed primarily against the cytoplasmic protein antigens proteinase 3 (PR3) and myeloperoxidase. On indirect immunofluorescence, these antigens adopt a fluoroscopic pattern that is either perinuclear (p-ANCA), cytoplasmic (c-ANCA), or atypical. The atypical pattern (x-ANCA or a-ANCA) includes features common to both. Although most c-ANCA recognize PR3 and most p-ANCA recognize myeloperoxidase, a percentage of p-ANCA are directed against other components of primary cytoplasmic granules, such as elastase and cathepsin, or components of the secondary granules, such as lactoferrin.10 These antibodies activate and trigger the degranulation of polymorphonuclear cells, promoting their adhesion to the endothelial cells and the generation of reactive oxygen species. However, a positive ANCA test result should be interpreted with caution because the presence of ANCA is also associated with infectious diseases (malaria and human immunodeficiency virus [HIV]), gastrointestinal diseases (inflammatory bowel disease, autoimmune hepatitis, primary biliary cirrhosis), and connective tissue diseases (lupus erythematosus, rheumatoid arthritis), and they are occasionally found in healthy individuals.11
Other factors that may be related to the pathophysiology of systemic vasculitis are antiendothelial cell antibodies, which are also involved in the development of other autoimmune connective tissue diseases through direct and indirect action on the vascular endothelium.12 Furthermore, a number of authors have described genetic polymorphisms that are associated, to a greater or lesser degree, with an increased risk of developing autoimmune diseases. Two examples are the case of the CD18 gene, associated with microscopic polyangiitis and the Churg–Strauss syndrome,13 and human leukocyte antigens A2, A11, and B35 alleles, associated with Henoch–Schönlein purpura.14
Clinical CharacteristicsThe different types of vasculitis give rise to a variety of primary lesions: the morphology of these lesions depends on vessel size, the anatomical site affected, and the stage of development of the lesion.
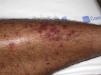
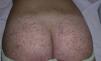
The most common primary lesions are purpuric macules or papules (palpable purpura) (Fig. 1) secondary to the involvement of the small caliber blood vessels of the skin. Other lesions that may occur include hemorrhagic blisters, pustules, urticarial or annular plaques, and nodules of varying depths that can become ulcerated. The nodules may be associated with livedo reticularis and represent damage to the vessels of the deep dermis and hypodermis. All of these lesions are found more frequently on the lower limbs, probably owing to hemodynamic factors. Cutaneous involvement in sites other than the lower limbs and the presence of nodular lesions, livedo reticularis, and ulcers are indications of vasculitis with systemic involvement. Irrespective of the underlying process or trigger, any vasculitis of the skin may be accompanied by fever, fatigue, or joint pain.15
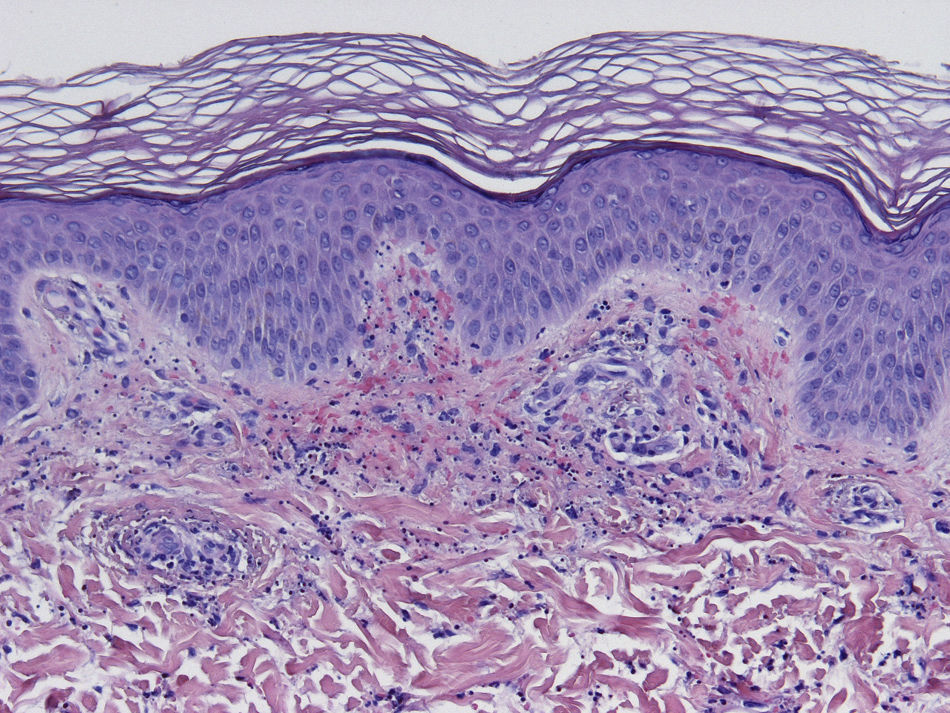
Histopathology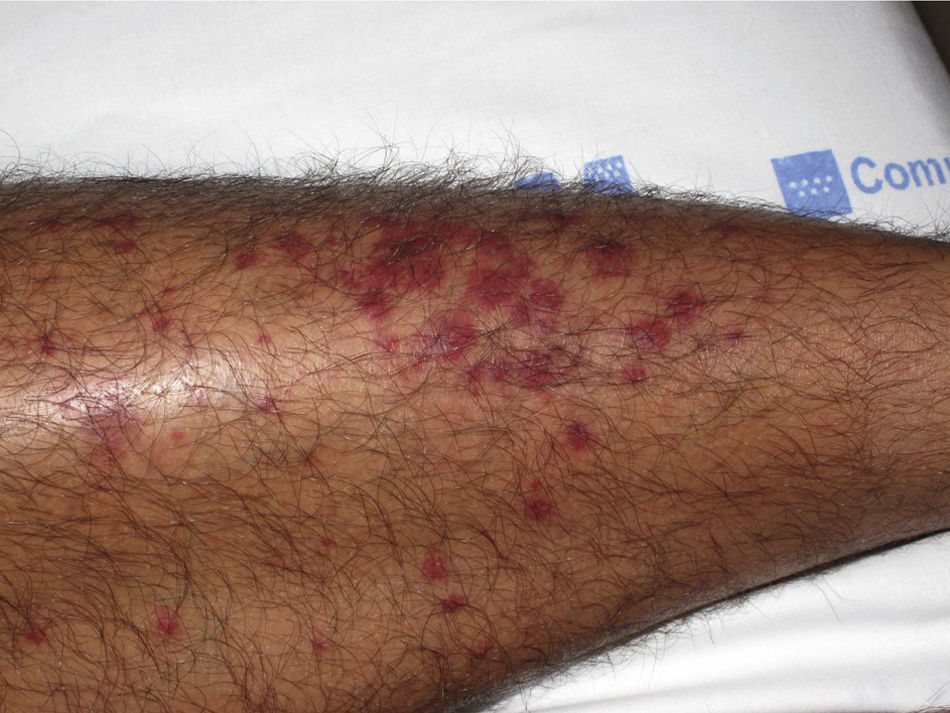
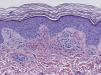
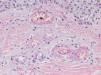
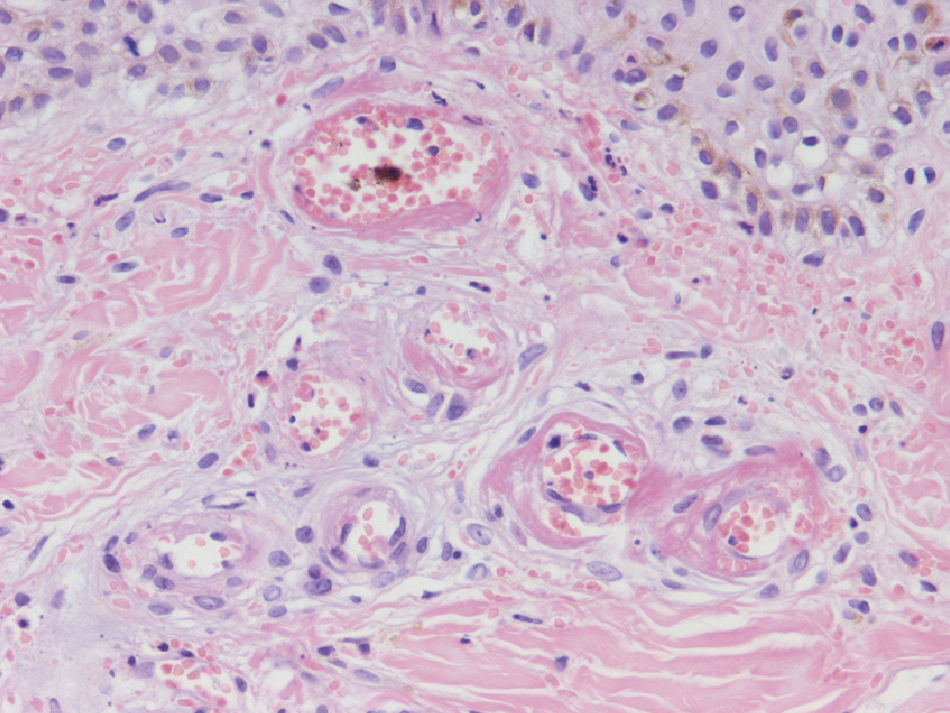
Since the term vasculitis corresponds to a purely histologic concept, skin biopsy is essential in the management of suspected vasculitis (Table 2). Biopsy samples should be obtained from fresh, nonulcerated lesions that have appeared within the preceding 48hours. The type of vessel involved (small, medium, or large) and the nature of the inflammatory infiltrate (neutrophilic, lymphocytic, or granulomatous) are the main factors taken into account in the histologic study. Neutrophils are found in most cases of vasculitis related to infections, toxic agents, drugs, food, autoimmune connective tissue diseases, type II and III cryoglobulins, and disease mediated by immunoglobulin (Ig) A. The disintegration of these neutrophils (leukocytoclasis) gives rise to nuclear dust (Fig. 2). However, in some types of vasculitis, the predominant cell type can be lymphocytes or monocytes. Infiltrate content will depend on the timing of the skin biopsy because polymorphonuclear leukocytes may be replaced by lymphocytes and monocytes as the lesions develop. In addition, the presence of the characteristic features of the different types of vasculitis should be assessed, for example the presence of extravascular granulomas. Edema and inflammatory infiltration of the vessel wall lead to progressive thickening of the wall, and very slight fibrinoid necrosis of the vessel walls will sometimes be observed (Fig. 3). Endothelial cells are frequently swollen and intravascular thrombi or extravasation of red blood cells into the dermis may occasionally be observed (Table 2). Marked subepidermal edema sometimes gives rise to vesiculobullous skin lesions. Direct immunofluorescence examination will provide additional information in the assessment of vasculitis. It should be performed on lesional skin and will reveal deposition of IgM, fibrinogen, and C3 in recent lesions, IgG in completely developed lesions, and fibrinogen and C3 in old lesions. The presence of IgA deposits may be more evident in patients at risk for renal failure.16
Histopathologic Criteria in the Diagnosis of Vasculitis.
| Histologic definition: inflammatory process affecting the vessel wall |
| Two criteria must be met: |
| 1. Inflammatory infiltrate in the vessel wall |
| 2. Damage in the form of fibrin deposition and/or endothelial necrosis |
| Histopathologic criteria for vasculitis |
| Major |
| Neutrophils and karyorrhexis (nuclear dust) |
| Necrosis and fibrin deposition |
| Minor |
| Endothelial cell swelling |
| Hemorrhage |
| Thrombosis |
| Necrosis |
| Epidermal vesicles |
| Histiocytes |
| Eosinophils |
| Aneurysms |
| Calcinosis |
We describe below some of the different types of vasculitis with cutaneous manifestations that constitute separate entities and briefly summarize the main clinical and histopathologic characteristics of each one.
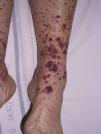
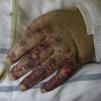
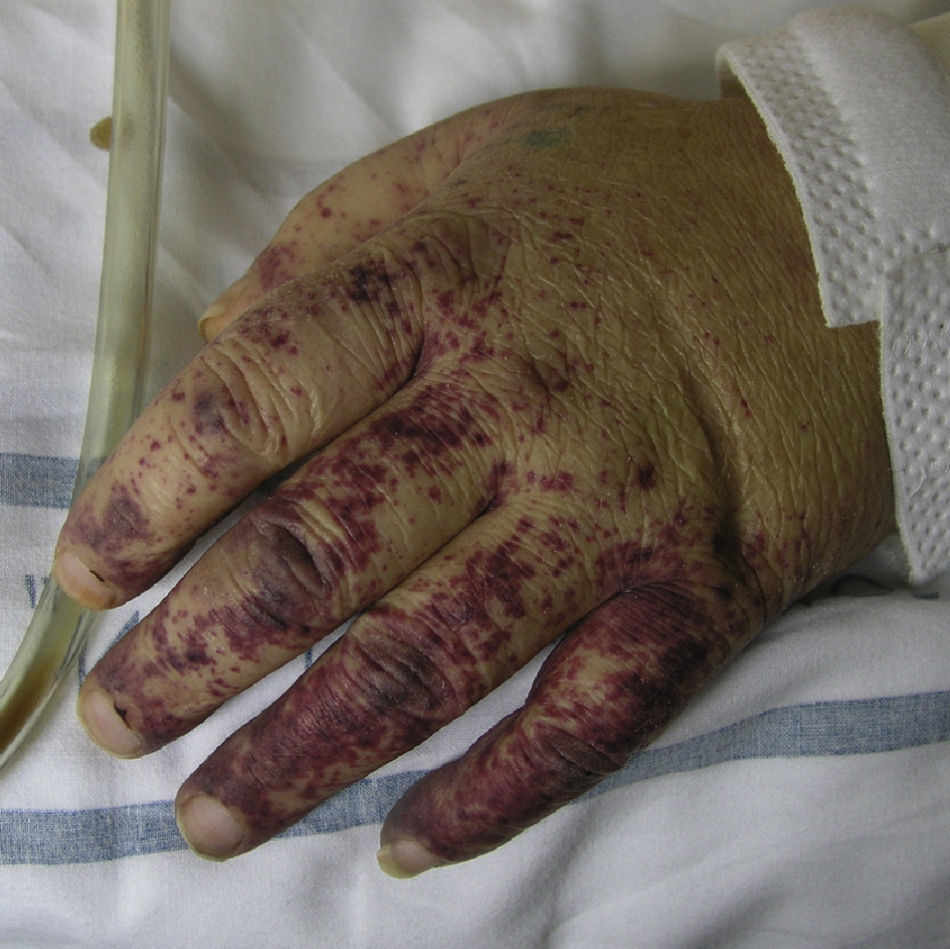
Types of Vasculitis That Constitute Separate EntitiesSmall Vessel Leukocytoclastic VasculitisSmall vessel leukocytoclastic vasculitis is the most common type of vasculitis encountered in dermatologic practice. It has also been called hypersensitivity vasculitis and cutaneous necrotizing venulitis. The clinical manifestations are purpuric lesions and, occasionally, erythematous papules, vesicles, blisters, pustules, or annular plaques; lesions are generally located on the lower limbs or in the dependent areas of the body (Fig. 4). This clinical picture may sometimes be preceded by or associated with extracutaneous symptoms such as arthralgia, fatigue, fever, or anorexia. While this entity generally affects only the skin, the same kind of skin lesions can occur in other forms of vasculitis that do have systemic involvement. Histopathological analysis reveals involvement of dermal postcapillary venules, with polymorphonuclear infiltration of the vessel walls and intravascular and extravascular eosinophils.17
The factors associated with the development of small vessel cutaneous vasculitis are shown in Table 3.18,88 The specific origin of the condition is never determined in up to 50% of patients. Some of these cases are associated with the presence of ANCA, and particularly antimyeloperoxidase or p-ANCA, usually in association with the administration of drugs such as hydralazine or propylthiouracil.19
Etiology of Cutaneous Small Vessel Leukocytoclastic Vasculitis.a
| Associated with precipitating factors | ||
| – | Infections | |
| + | Bacterial | |
| =Streptococcus | ||
| =Staphylococcus | ||
| =Meningococcus | ||
| =Gonococcus | ||
| =Pseudomonas | ||
| =Treponema pallidum | ||
| =Mycobacterium leprae | ||
| =Mycobacterium tuberculosis (?) | ||
| + | Viral | |
| =Hepatitis B | ||
| =Influenza | ||
| =Cytomegalovirus | ||
| + | Parasitic | |
| =Plasmodium | ||
| – | Drugs | |
| + | Penicillins | |
| + | Tetracyclines | |
| + | Sulfonamides | |
| + | Erythromycin | |
| + | Griseofulvin | |
| + | Anti-inflammatory agents (aspirin, phenacetin) | |
| + | Thiazides | |
| + | Loop diuretics | |
| + | Propylthiouracil | |
| + | Penicillamine | |
| + | Phenothiazines | |
| + | Quinidine | |
| – | Chemical substances | |
| + | Insecticides | |
| + | Herbicides | |
| + | Petroleum derivatives | |
| Associated with chronic persistent disorders | ||
| – | Autoimmune connective tissue diseases | |
| + | Systemic lupus erythematosus | |
| + | Rheumatoid arthritis | |
| + | Sjögren syndrome | |
| – | Inflammatory bowel diseases | |
| + | Ulcerative colitis | |
| + | Crohn disease | |
| – | Mixed cryoglobulinemia | |
| – | Hypergammaglobulinemic purpura of Waldenström | |
| Associated with neoplasms | ||
| – | Hematologic neoplasms | |
| – | Solid tumors | |
| Idiopathic or unclear cause | ||
Any viral, bacterial, parasitic, or fungal infection can cause cutaneous vasculitis. The pathogenesis of the disease probably involves a combination of the following pathophysiological mechanisms: the formation of immune complexes (hepatitis B or C virus, or HIV), direct endothelial damage (cytomegalovirus), direct activation of the complement system (Candida species), or the formation of autoantibodies.20 Paraneoplastic vasculitides, which are mainly associated with hematologic neoplasms, may be the result of direct invasion of the vessel wall, the formation of immune complexes, or direct stimulation of cellular immunity by tumor antigens.21
Cutaneous vasculitis can occur in the context of a number of different inflammatory autoimmune diseases, including systemic lupus erythematosus,22,23 rheumatoid arthritis,24 Sjögren syndrome,25 as well as inflammatory bowel disease.26
Finally, there is a group of cutaneous vasculitides secondary to contact with chemicals (insecticides, petroleum derivatives, or topical drugs27) and plant or animal substances, or associated with eating certain foods or additives.28
Henoch–Schönlein PurpuraHenoch–Schönlein purpura, also known as anaphylactoid purpura and purpura rheumatica, is the most common subtype of vasculitis in childhood, with peak incidence at 7 years of age. While the condition affects both sexes equally in childhood, there is a slight male preponderance in adults. Although in some cases this condition occurs after a streptococcal infection of the upper airways, other factors have also been implicated in its pathogenesis, including other bacteria (Staphylococcus aureus, Campylobacter jejuni, Mycoplasma pneumoniae29), certain viruses (hepatitis A and B, adenovirus, coxsackie virus, parvovirus B1930), parasites (Toxocara canis31), drugs, vaccines, and even insect bites.32 IgA appears to play a key role in the pathogenesis of this entity, as evidenced by the presence of IgA immune complexes in dermal vessel walls and the glomerular mesangium. Specifically, it appears that aberrant glycosylation of IgA1 determines its affinity for forming circulating immune complexes and depositing them in these areas.33
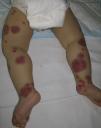
Clinically, Henoch–Schönlein purpura is characterized by the presence of signs and symptoms affecting the skin, gastrointestinal system, joints, and kidneys. Skin lesions initially appear as macules or papules with an urticarial appearance, which subsequently progress to inflammatory and purpuric papules. Other, less common presentations include vesicles, blisters, and focal areas of necrosis. Lesions are usually distributed symmetrically across both lower limbs and buttocks (Fig. 5), although in some cases they may extend up to the trunk or as far as the arms. Gastrointestinal or musculoskeletal involvement is observed in up to 75% of patients. In such cases, the manifestations include nausea, vomiting, abdominal pain, gastrointestinal bleeding, and arthritis of large joints (for example, the ankles and knees).34,35 Leg edema is not uncommon. Renal involvement occurs in up to 50% of cases and is expressed as hematuria (macroscopic or microscopic), proteinuria, and nephrotic or nephritic syndrome. Nephropathy progresses to terminal renal failure in between 1% and 3% of these patients. Orchitis, intussusception, pancreatitis, uveitis, neurologic complications, and pulmonary hemorrhage have also been reported.36
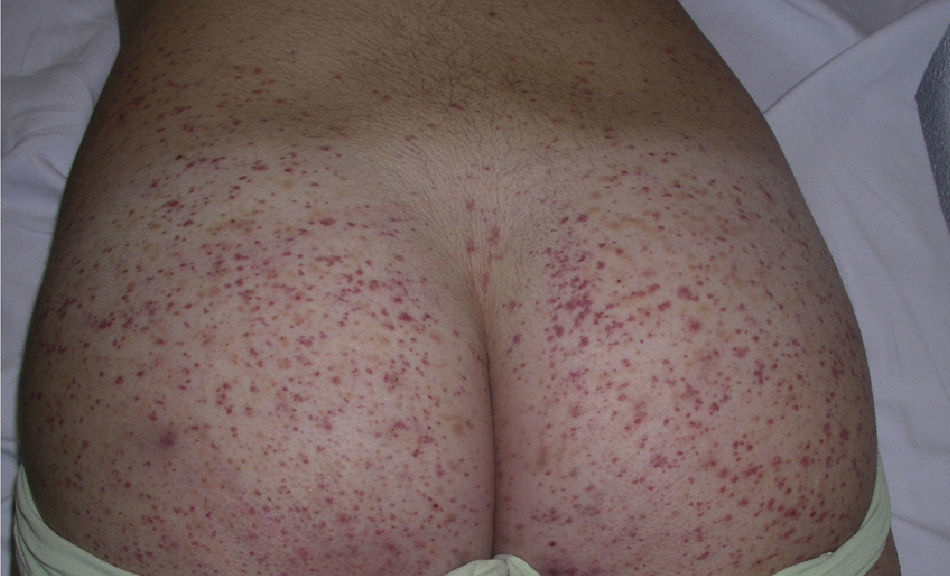
Histopathological findings with conventional stains demonstrate the presence of a leukocytoclastic vasculitis of the small blood vessels in the dermis. If the skin biopsy is recent (under 48h), IgA deposits (usually IgA1) can be observed in the vessel walls.
There are no laboratory test findings characteristic or pathognomonic of the disease, although the concentration of acute-phase reactants is usually elevated. An increase in circulating polyclonal IgA is observed in 25% to 50% of cases.
Diagnosis is achieved by correlating clinical and histopathological findings, and special consideration should be given to the findings of the direct immunofluorescence study. However, it should never be forgotten that a diagnosis of Henoch–Schönlein vasculitis is not synonymous with IgA mediated vasculitis.
Urticarial VasculitisUrticarial vasculitis is characterized by the presence of wheals that persist for more than 24h. The condition was initially called urticariform vasculitis by McDuffie37 and hypocomplementemic vasculitis (although cases were later reported in which circulating complement levels were not reduced).38 The pathogenesis of the hypocomplementemic forms has been linked to the presence of anti-C1q antibodies, a finding that has led some authors to consider that they should be included within the spectrum of systemic lupus erythematosus.39,40 The onset of urticarial vasculitis has also been linked to other connective tissue diseases (Sjögren syndrome41) as well as infections (hepatitis B and infectious mononucleosis), drugs,42 IgM and IgA paraproteinemia, serum sickness-like reaction, and certain tumors.43
Clinically, urticarial vasculitis manifests as wheals, which typically contain purpuric foci at some time during their development; these lesions are often accompanied by angioedema. The hypocomplementemic form may be associated with systemic symptoms affecting the joints, eyes, lungs, or digestive system. Classically, it has been assumed that the histopathologic findings would be compatible with small vessel leukocytoclastic vasculitis of the skin accompanied by marked edema in the upper dermis. However, some authors have reported a predominance of neutrophils in the lesions of the hypocomplementemic forms, while in the normocomplementemic variants vasculitis is lymphocytic and accompanied by a variable number of eosinphils.44
Erythema Elevatum DiutinumEythema elevatum diutinum is classified as a neutrophilic dermatosis and is characterized by the presence of symmetrical lesions on the backs of the hands and the extensor surfaces of the limbs.45 One of the main factors involved in its pathogenesis is the deposition of circulating immune complexes.46 Eythema elevatum diutinum has been associated with infections (group A β-hemolytic streptococcus, HIV, and hepatitis B virus), autoimmune diseases (systemic lupus erythematosus, inflammatory bowel disease, rheumatoid arthritis, relapsing polychondritis), blood disorders (myelodysplastic syndrome, myeloproliferative syndromes, multiple myeloma, hairy cell leukemia), and certain solid tumors.47 The clinical presentation includes papules, and violaceous, yellowish, or erythematous plaques or nodules. Extracutaneous involvement is exceptional, although some patients report pain in the joints underneath skin lesions. Histopathologic findings vary according to the timing of the biopsy. Lesions of recent onset display leukocytoclastic vasculitis of the dermal small vessels accompanied by perivascular neutrophilic infiltrate of variable intensity. In more developed lesions, the neutrophils occupy the entire dermis and lymphocytes, histiocytes, and eosinophils are also be found. Since erythema diutinum, like granuloma faciale, is a form of chronic fibrosing leukocytoclastic vasculitis, old lesions will display a variable degree of perivascular and dermal fibrosis.48 Extracellular cholesterolosis, which refers to the deposition of cholesterol in the extracellular spaces and in the histiocytes of long-standing lesions,49 is now rarely seen because of the introduction of dapsone for the treatment of this condition.
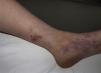
Acute Hemorrhagic Edema of InfancyAcute hemorrhagic edema of infancy, also known as Finkelstein disease and Seidlmayer syndrome, is a benign condition only observed in infancy. Typically, it develops during the course of or after the resolution of urinary or respiratory tract infection, following the administration of certain vaccines, or during treatment with certain drugs. Like other immune-mediated forms of vasculitis, it is probably caused by immune complexes that form in response to an infectious or pharmacologic antigenic stimulus.50 The condition is characterized by the sudden appearance of bruise-like erythmatopurpuric plaques with a nummular or cockade appearance on the face or distal areas of the limbs (Fig. 6); these lesions may become vesiculobullous or necrotic. Extracutaneous involvement is extremely rare. This type of vasculitis is uncommon in patients over 2 years of age. Histopathologic analysis reveals dermal edema in association with small vessel leukocytoclastic vasculitis. IgA deposition in the vessel walls is observed in up to one third of cases, leading some authors to consider that this condition could be a subtype of Henoch–Schönlein purpura.51 While this is a benign condition that usually requires no treatment, it is essential to rule out processes such as exudative erythema multiforme, Kawasaki disease, meningococcemia, Sweet syndrome, and even child abuse.
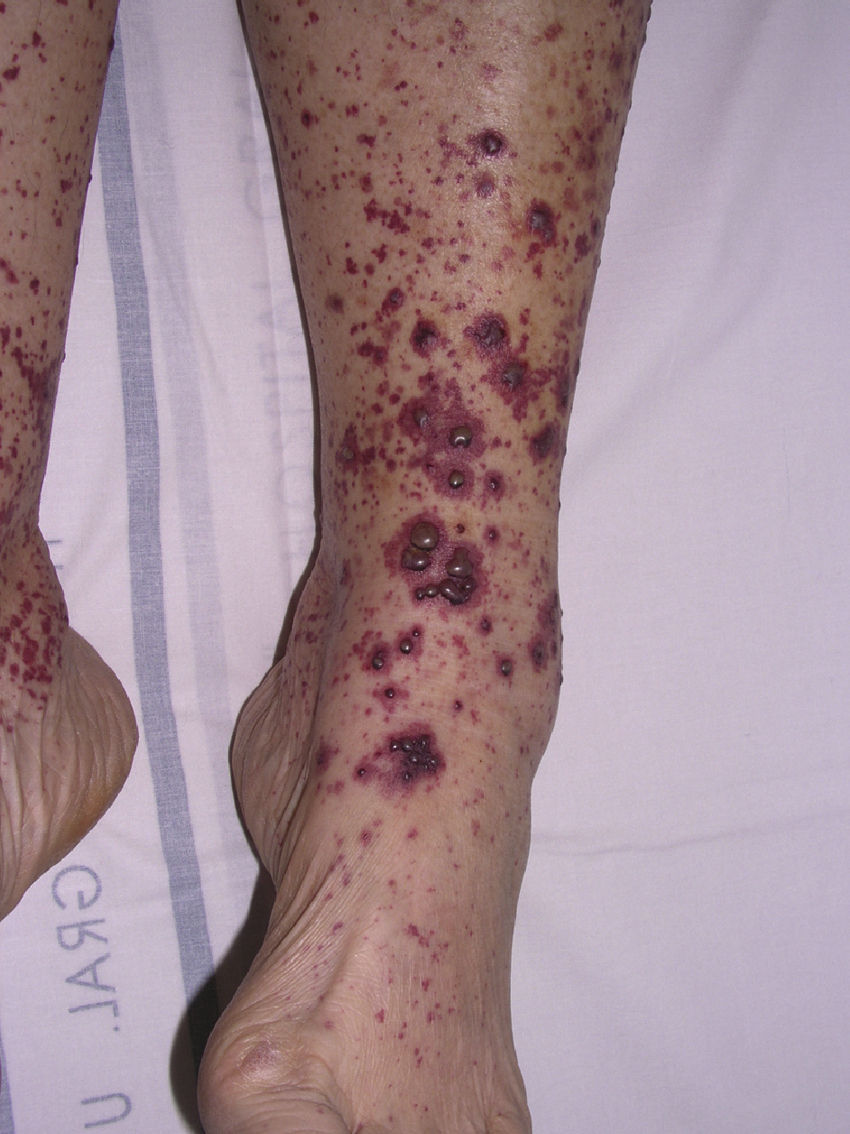
Cryoglobulinemic Vasculitis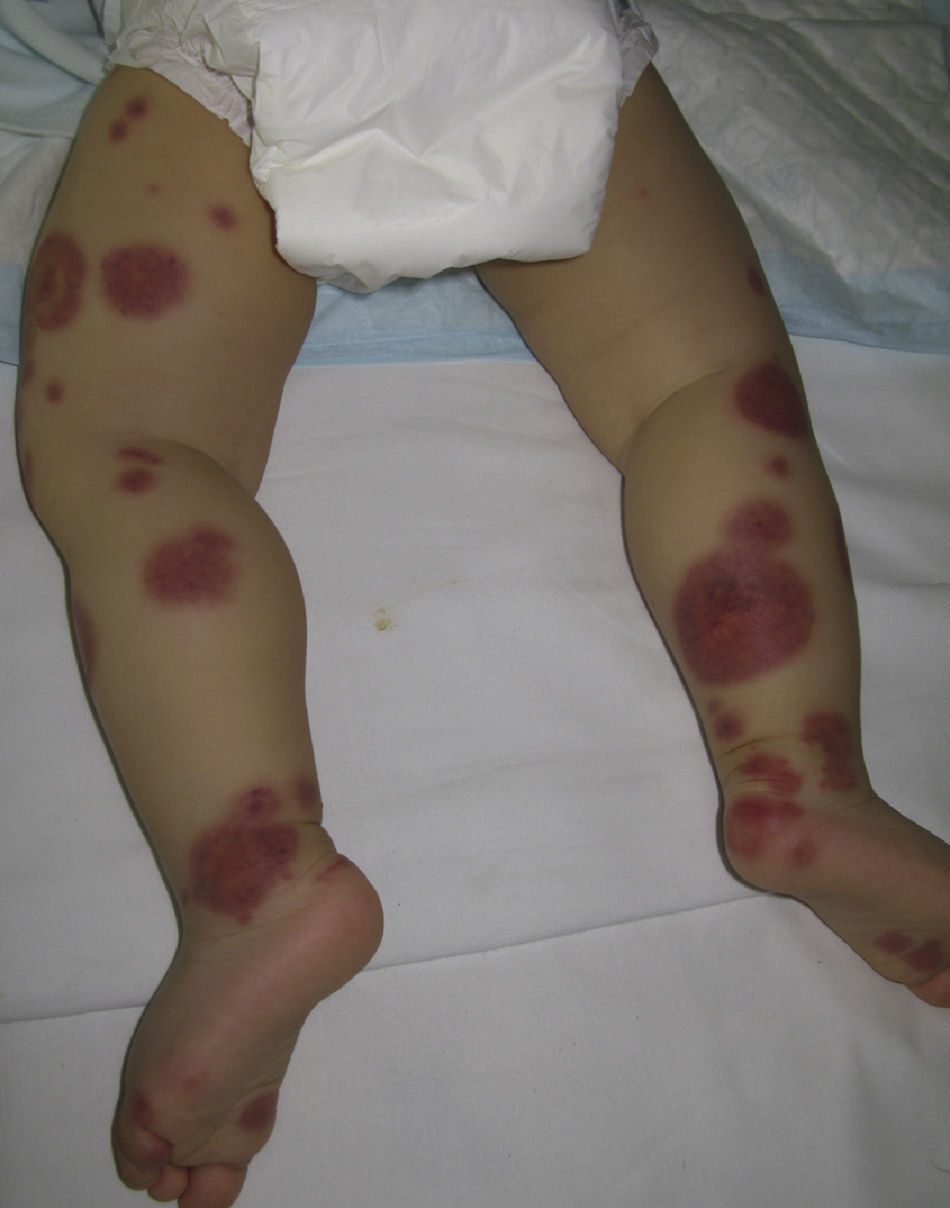
Cryoglobulinemic vasculitis affects small and medium-sized vessels and is caused by the deposition in the vessel walls of circulating immune complexes formed by cryoglobulins.52 These immunoglobulins precipitate at temperatures below 37°C, dissolving again once the temperature rises above this level. Based on the immunochemical composition of the cryoglobulins present we can distinguish 2 types of cryoglobulinemia: monoclonal and mixed. The monoclonal form is associated with the presence of a monoclonal immunoglobulin (IgG or IgM) and no rheumatoid factor activity; these IgG and IgM complexes are classified as type I cryoglobulins. Most patients with this variant have an underlying hematologic disorder (multiple myeloma, Waldenström macroglobulinemia, or chronic lymphocytic leukemia), which presents as purpura, acral cyanosis, ulcers, or livedo reticularis. Microscopy reveals eosinophilic thrombi composed of a period-acid-Schiff-positive material that may or may not be accompanied by a perivascular inflammatory infiltrate composed of lymphocytes. In the mixed variant, the cryoglobulins may comprise either a polyclonal (IgG) and a monoclonal (IgM) component with rheumatoid factor activity (type II cryoglobulins) or a number of polyclonal IgG and IgM molecules with rheumatoid factor activity (type III cryoglobulins).53 The mixed forms are usually associated with hepatitis C infection, autoimmune diseases such as Sjögren disease, and lymphoproliferative processes. However, in a not insignificant percentage of patients no causal agent is identified and the condition is called idiopathic or essential mixed cryoglobulinemia. Clinically, cryoglobulemic vasculitis is characterized by the presence of predominantly acral purpuric lesions (Fig. 7), which are sometimes associated with ulcers or areas of necrosis. Most patients report worsening of symptoms with exposure to cold. Histopathologic analysis reveals leukocytoclastic vasculitis affecting the small and medium-sized vessels of the dermis and hypodermis. The presence of intravascular hyaline thrombi is a rare finding, usually observed near ulcerated areas. Direct immunofluorescence reveals deposition of immunoglobulins and C3 in the vessel walls. Other organs and tissues commonly affected are the kidneys (membranoproliferative glomerulonephritis), the peripheral nervous system, the joints, and, to a lesser degree, the lungs.54
Measurement of cryoglobulin levels in serum may give rise to false negatives due to the influence of the ambient temperature on their solubility and the possible decline in such levels during the intercritical period. It should be remembered that the presence of circulating cryoglobulins is not always associated with symptoms. The term cryoglobulinemic syndrome is only used to describe symptomatic cases. A high percentage of patients have elevated plasma concentrations of rheumatoid factor and decreased levels of complement C4.55
The prognosis in these patients basically depends on the extent of renal involvement, the presence or absence of a concomitant tumor, or—especially in the mixed forms linked to hepatitis C virus—progression to a lymphoproliferative process.56
Septic VasculitisThe septic vasculitides are a subgroup of acute vasculitis associated with septicemia. Patients with acute meningococcemia develop irregularly shaped purpuric lesions; findings include a perivascular and interstitial neutrophilic inflammatory infiltrate, and focal areas of leukocytoclasis, fibrinoid necrosis, and vessel occlusion.57 Gram staining reveals the presence of abundant gram-negative diplococci in endothelial cells and polymorphonuclear leukocytes. Immediate treatment with antibiotics is essential, although blood cultures are needed to confirm the diagnosis. The chronic forms of meningococcemia and gonococcemia are characterized by a triad of intermittent fever, arthralgia, and vesiculopustular or purpuric skin lesions. The histopathologic features differ slightly from those of the classic forms of hypersensitivity vasculitis, and include arteriole involvement, a predominance of neutrophils, and the formation of subepidermal and intraepidermal pustules. Usually, no organisms are visible on gram staining, although bacteria have been identified in some cases by molecular genetic techniques.58 Blood cultures are positive during the febrile phase. Treatment with systemic corticosteroids can lead to severe complications.59
Wegener GranulomatosisWegener granulomatosis is a rare disorder characterized by involvement of the upper and lower airway and the renal glomerulus secondary to the presence of systemic vasculitis and focal areas of granulomatous and necrotic inflammation. While this disorder affects a wide range of ages, onset usually occurs in patients between 40 and 50 years of age. Its pathogenesis is poorly understood, but the presence of an amplified immune response to a specific antigenic stimulus could be one of the mechanisms involved. One of the stimuli suggested as a possible trigger for this breakdown of immune tolerance is infection with S. aureus. In addition to the involvement of c-ANCA, there is growing recognition of the role played by T lymphocytes in the pathogenesis of this disorder.60 Typically, patients initially present with upper airway symptoms (rhinitis, sinusitis, ulceration of the nasal septum) or report nonspecific respiratory symptoms such as dyspnea or a persistent dry cough. However, up to 10% of patients may present with hemoptysis or alveolar hemorrhage. Renal involvement is common and irreversible glomerular damage is one of the factors that determines a poorer prognosis. The presence of cutaneous manifestations is variable and skin lesions are the presenting symptom in under 10% of patients. The most common skin lesion is purpura on the lower limbs, although a more specific sign is papulonodular lesions with central necrosis distributed symmetrically on the trunk and limbs. The second most common finding on examination is mucocutaneous ulceration, which usually takes the form of hyperplastic or strawberry-like gingivitis.61 Pertinent laboratory findings include elevations in white blood cell count, activated protein C, and erythrocyte sedimentation rate. In approximately 80% of patients, c-ANCA specific to the PR3 antigen are found; the usefulness of PR-3 antigen titers as a predictor of risk of recurrence after treatment is still a matter of debate.62,63 Typically, histopathologic analysis reveals the presence of a triad comprising vasculitis, areas of necrosis, and granulomatous inflammation. The most common finding in lesional skin biopsies is a neutrophilic vasculitis of the small and medium-sized vessels. A small number of patients present with a granulomatous inflammatory infiltrate composed of macrophages, neutrophils, and multinucleated giant cells surrounding areas of necrosis. In some studies, the presence of cutaneous vasculitis correlated with increased disease activity and progression to renal involvement.64 The clinical course of the disease is marked by recurrence, observed in up to half of all patients.
Microscopic PolyangiitisMicroscopic polyangiitis, once considered a subtype of polyarteritis nodosa, is now classified as a separate entity. It is a small and medium-sized vessel vasculitis with systemic involvement. The organs most often affected are the kidneys (79%–90%) with rapidly progressive glomerulonephritis and the lungs (25%–50%) in the form of pulmonary hemorrhage.65,66 This disorder is usually preceded by a prodromal phase characterized by myalgia, arthralgia, fever, and weight loss. Occasionally, patients may present gastrointestinal symptoms or signs of peripheral neuropathy. The cutaneous lesions are indistinguishable from those of small vessel cutaneous leukocytoclastic vasculitis. In up to 70% patients, p-ANCA are found. Histopathologic analysis of samples of purpuric skin lesions demonstrates small vessel leukocytoclastic vasculitis, but a direct immunofluorescence assay does not reveal immune complex deposition.67 A number of viruses have been implicated in the etiology and pathogenesis of this disorder (hepatitis B and C virus), although, as in other types of vasculitis, the underlying causal mechanism is poorly understood.
Churg–Strauss SyndromeChurg–Strauss syndrome, also known as allergic granulomatosis, is a type of systemic vasculitis characterized by the association of persistent severe asthma and eosinophilia in peripheral blood. This disease predominantly affects those aged between 30 and 50 years and affects both sexes equally. From onset in adults, respiratory symptoms range from allergic rhinitis, nasal polyposis, and pansinusitis to corticosteroid-dependent asthma. Response to conventional asthma treatments is usually poor. Triggering factors include certain vaccinations, desensitization processes, and abrupt withdrawal of corticosteroid treatment. There is still no consensus on the possible role in this setting of leukotriene receptor antagonist therapy or monoclonal anti-IgE antibodies such as omalizumab.68,69 Churg–Strauss syndrome evolves in several clinical stages: the first involves respiratory symptoms and peripheral eosinophilia; and in the second, disease affects the lungs and heart as well as the digestive, musculoskeletal, and nervous systems. Skin involvement can occur at onset or, in about 50% of patients, develop at a later stage. The most common cutaneous manifestations are purpuric macules or papules on the lower limbs. Another common finding in these patients is the presence of intact nodules (which may progress to ulceration) on the scalp and the distal areas of the limbs. Cases have also been reported in which the predominant cutaneous manifestations were livedo racemosa, urticarial lesions, or vesiculopustular lesions.70 Pathologic examination can reveal, although not often simultaneously, 3 characteristic features: small and medium vessel leukocytoclastic vasculitis, tissue eosinophilia, and an extravascular granulomatous inflammatory infiltrate composed of a large number of eosinophils and their degradation products (red granulomas). Blood analysis shows intense eosinophilia, which is accompanied by increased IgE during flares. As many as half of all patients test positive for p-ANCA, but c-ANCA is found in only 10% to 20%. In a study by Sable-Fourtassou and colleagues,71 the presence of ANCA correlated with a higher probability of finding evidence of vasculitis in the histopathologic study.
Traditionally, the prognosis for patients with Churg–Strauss vasculitis was poor: overall survival was less than 1 year and the leading cause of death was heart failure. Today, as a result of prompt treatment with corticosteroid and immunosuppressive therapy, prolonged remission can be achieved in most cases.72
Polyarteritis NodosaPolyarteritis nodosa is a vasculitis that affects small and medium-sized arteries, usually resulting in damage to multiple organs. Incidence is highest between 40 and 60 years of age and is related to vaccinations or infection with hepatitis B,73 hepatitis C, HIV, or parvovirus B19.74 Clinical signs vary depending on the organ or tissue affected, but this entity has a predisposition for the peripheral nervous system (polyneuritis), the digestive system (gastrointestinal bleeding), and the renal vasculature (hypertension and renal infarction). Cutaneous symptoms, which may indicate disease onset, include purpuric nodules that tend to ulcerate, livedo reticularis, and necrotic lesions (Fig. 8). Histopathologic findings in lesional skin can vary owing to segmental damage to the vascular wall. Typically, the small vessels of the dermis and arterioles located in the subcutaneous tissue are affected. In the early stages, a neutrophilic inflammatory infiltrate predominates, usually extending into subcutaneous tissue and giving rise to a microscopic appearance of lobular panniculitis. These neutrophils are later replaced by a variable number of lymphocytes. Progressive obliteration of vascular lumens is usually an indication of microaneurysms. Laboratory test findings include the presence of an inflammatory syndrome with an elevated erythrocyte sedimentation rate and, occasionally, eosinophilia. ANCA are detected in a small proportion of patients (10%–20%), usually distributed in a perinuclear pattern.
A localized form of polyarteritis nodosa exists that predominantly affects the skin. This variant, called cutaneous polyarteritis nodosa, is a chronic recurrent disorder characterized by the presence of nodular lesions and livedo racemosa particularly on the distal third of the lower limbs—symptoms similar to those described for the classic form of the disease. The lesions may spread to other limbs and outbreaks are sometimes accompanied by myalgia, fatigue, edema, and fever.
DiagnosisOnce the diagnosis has been confirmed by skin biopsy findings, the extent of disease should be assessed. The first step is to obtain a complete medical history and perform a thorough physical examination, paying particular attention to the heart, joints, and respiratory, digestive, and nervous systems. Blood analysis and urine sediment, electrocardiogram, and a chest radiograph should be obtained. Etiology is established by correlating clinical signs and laboratory findings specific to or suggestive of particular entities. Therefore, in cases of recurrence or when systemic involvement is likely, the workup should be expanded to identify the possible presence in blood of antinuclear antibodies, ANCA, rheumatoid factor, cryoglobulins, total complement and its fractions, and antistreptococcal antibodies; immunoelectrophoresis of proteins should also be performed. In selected cases, additional diagnostic procedures may be required to determine the involvement of specific organs or tissue (for example, renal or nerve biopsy, bone marrow aspiration, or selective arteriography) (Fig. 9).75–77
Differential Diagnosis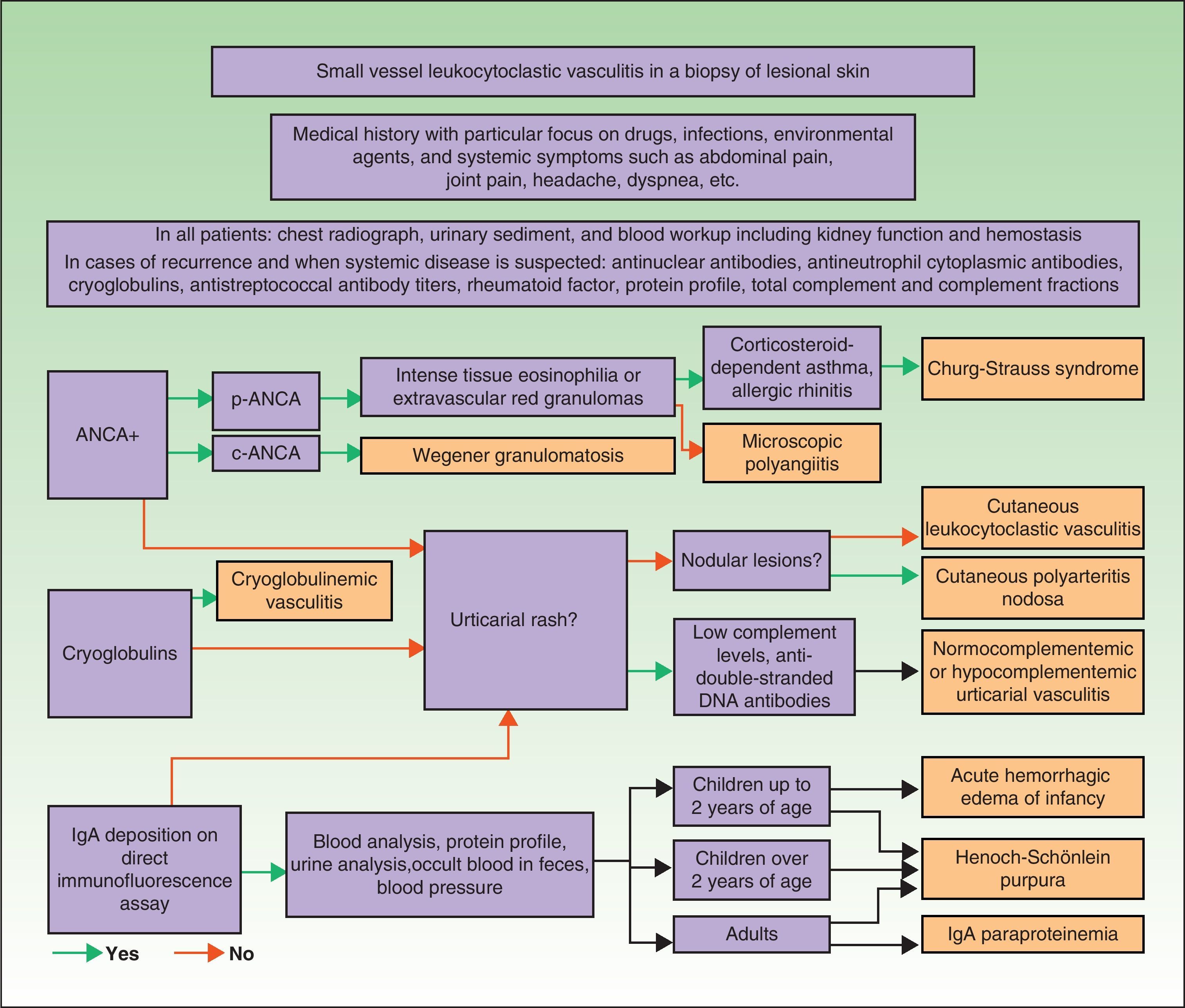
The possible implication of nonvasculitic processes must also be taken into account when investigating the possible cause of mucocutaneous purpura. Capillary fragility is seen in a large proportion of elderly or corticosteroid-dependent patients and in individuals with vitamin C deficiency78 or primary systemic amyloidosis.79 The pigmented purpuras are a group of benign dematoses, usually of unknown etiology (Schamberg disease, lichen aureus, eczematid-like purpura, and purpura annularis telangiectodes or Majocchi disease) or associated with chronic venous insufficiency (Favre–Chaix angiodermatitis, purpuric dermatitis, and purpuric lichenoid dermatitis of Gougerot and Blum).80 The possibility of purpura having a primarily thrombotic origin must also be considered in the differential diagnosis of the types of vasculitis. For example, thrombosis is involved in antiphospholipid syndrome, disseminated intravascular coagulation, and C and S protein deficiencies. However, in patients who have undergone endovascular procedures, are polytraumatized or have cardioembolic disease, the presence of an underlying embolic process should be suspected.81,82 Other conditions that simulate vasculitis through diverse mechanisms that cause vascular occlusion include calciphylaxis, cholesterol embolism, hyperoxaluria, and hyperhomocysteinemia, among others.
TreatmentIf there are known predisposing or precipitating factors, the first concern of treatment should be to eliminate possible underlying causes.
With respect to the symptomatic treatment of skin lesions, we recommend bed rest with elevation of the lower limbs and treatment with nonsteroidal anti-inflammatory drugs (as long as these are not the cause of the symptoms) or antihistamines. Good results have also been achieved with colchicine83 at a dose of 0.5mg/8h and/or dapsone84 at a dose of 50–200mg/24h.
In cases of severe cutaneous vasculitis characterized by large necrotic or ulcerated areas, treatment with systemic corticosteroids is necessary; for example, prednisone in a regimen starting at a dose of 0.5mg to 1mg/kg/d and gradually tapering over 4–6weeks. If clinical relapse occurs when the dose is being reduced, corticosteroid-sparing immunosuppressive agents may be prescribed: for example, azathioprine (50–100mg/d) or methotrexate (10–25mg/wk).85
If there is systemic involvement, initial treatment should include high-dose corticosteroids and/or oral or intravenous cyclophosphamide pulse therapy until remission of symptoms is achieved. Treatment should be prescribed on a case-by-case basis taking into account the symptoms or entity being treated, the patient's renal status, and the specific contraindications that apply in each case.86 Once remission is achieved, a maintenance regimen can be instituted with azathioprine, mycophenolate mofetil, or methotrexate. Intravenous immunoglobulin or plasmapheresis can be useful in selected cases. Drugs such as infliximab and rituximab have shown promising results in the treatment of systemic vasculitis refractory to conventional therapies.87
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Pulido-Pérez A, et al. Vasculitis cutáneas. Actas Dermosifiliogr. 2012;103:179–91.


