



La xantomatosis cerebrotendinosa (XCT) es una enfermedad hereditaria infrecuente causada por la mutación del gen CYP27A1. Es característica la aparición de xantomas en diferentes tejidos, principalmente en el cerebro y los tendones, secundarios al depósito de β-colestanol. El diagnóstico se confirma mediante la determinación de β-colestanol en suero, y de los alcoholes biliares en orina. El ácido quenodesoxicólico es la terapia más eficaz, pudiendo llegar a frenar la progresión de la enfermedad. Presentamos 4 pacientes con alteraciones neurológicas desde la infancia que fueron diagnosticados de XCT tras el desarrollo de xantomas tendinosos. El reconocimiento de los xantomas tendinosos es fundamental para orientar el diagnóstico de XCT, pero estos no son un signo inicial de la enfermedad, que debuta con alteraciones neurológicas, cataratas o diarrea crónica. Por lo tanto, el diagnóstico temprano de la XCT requiere la determinación del β-colestanol sérico en estos pacientes, aun en ausencia de xantomas.
Cerebrotendinous xanthomatosis (CTX) is an uncommon autosomal recessive disease caused by mutation of the CYP27A1 gene. It is characterized by the presence of xanthomas in different tissues, principally brain and tendon, due to the accumulation of β-cholestanol. Diagnosis is confirmed by measurement of serum β-cholestanol and urinary bile alcohol levels. Therapy with chenodeoxycholic acid has been shown to be the most effective treatment and can halt progression of the disease. We present 4 patients with a history of neurological disorders since childhood and who were diagnosed with CTX after developing tendon xanthomas. Although diagnostic suspicion depends to a large extent on recognition of tendon xanthomas, these are not an early sign of the disease, which can present with neurological disorders, cataracts, and chronic diarrhea. Early diagnosis of CTX therefore rests on measurement of serum β-cholestanol levels, even in absence of tendon xanthomas.
La xantomatosis cerebrotendinosa (XCT) es una enfermedad autosómica recesiva producida por el déficit de la enzima 27α-esterol-hidroxilasa1,2. Las mutaciones del gen que codifica esta enzima (CYP27A1) son responsables de la enfermedad, caracterizada por la formación de xantomas en diferentes tejidos, sobre todo en el cerebro y los tendones3,4. El diagnóstico bioquímico se realiza mediante la demostración de niveles elevados de β-colestanol en suero y de alcoholes biliares en orina. El estudio molecular del gen permite el diagnóstico de los heterocigotos y el diagnóstico prenatal5,6. El diagnóstico temprano es fundamental para iniciar el tratamiento con ácido quenodesoxicólico (AQDC) y prevenir la progresión de la enfermedad en forma de deterioro neurológico. Presentamos 4 pacientes afectos de un cuadro neurológico desde la infancia, que fueron diagnosticados de XCT tras el desarrollo de xantomas tendinosos.
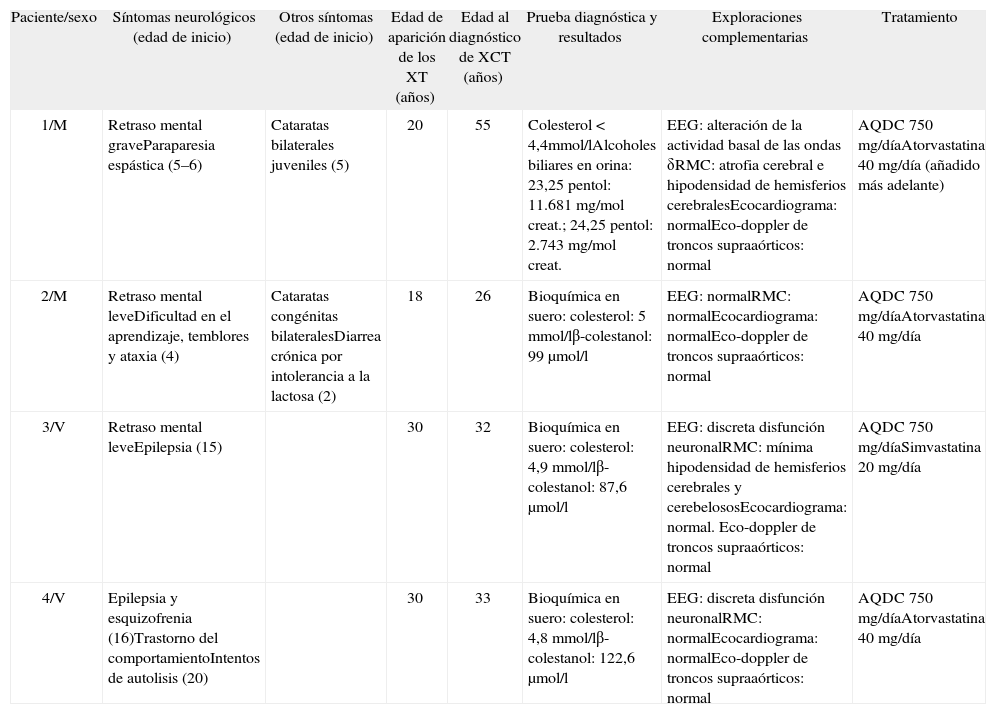
Casos clínicosPresentamos 4 pacientes, dos de ellos hermanos (pacientes 3 y 4), que acudieron a nuestro Servicio con edades comprendidas entre los 26 y los 55 años. La información clínica de los pacientes se resume en la tabla 1. El caso de la paciente 1 ya ha sido publicado previamente2.
Datos clínicos más relevantes de los 4 pacientes con xantomatosis cerebrotendinosa
| Paciente/sexo | Síntomas neurológicos (edad de inicio) | Otros síntomas (edad de inicio) | Edad de aparición de los XT (años) | Edad al diagnóstico de XCT (años) | Prueba diagnóstica y resultados | Exploraciones complementarias | Tratamiento |
| 1/M | Retraso mental graveParaparesia espástica (5–6) | Cataratas bilaterales juveniles (5) | 20 | 55 | Colesterol < 4,4mmol/lAlcoholes biliares en orina: 23,25 pentol: 11.681 mg/mol creat.; 24,25 pentol: 2.743 mg/mol creat. | EEG: alteración de la actividad basal de las ondas δRMC: atrofia cerebral e hipodensidad de hemisferios cerebralesEcocardiograma: normalEco-doppler de troncos supraaórticos: normal | AQDC 750 mg/díaAtorvastatina 40 mg/día (añadido más adelante) |
| 2/M | Retraso mental leveDificultad en el aprendizaje, temblores y ataxia (4) | Cataratas congénitas bilateralesDiarrea crónica por intolerancia a la lactosa (2) | 18 | 26 | Bioquímica en suero: colesterol: 5 mmol/lβ-colestanol: 99 μmol/l | EEG: normalRMC: normalEcocardiograma: normalEco-doppler de troncos supraaórticos: normal | AQDC 750 mg/díaAtorvastatina 40 mg/día |
| 3/V | Retraso mental leveEpilepsia (15) | 30 | 32 | Bioquímica en suero: colesterol: 4,9 mmol/lβ-colestanol: 87,6 μmol/l | EEG: discreta disfunción neuronalRMC: mínima hipodensidad de hemisferios cerebrales y cerebelososEcocardiograma: normal. Eco-doppler de troncos supraaórticos: normal | AQDC 750 mg/díaSimvastatina 20 mg/día | |
| 4/V | Epilepsia y esquizofrenia (16)Trastorno del comportamientoIntentos de autolisis (20) | 30 | 33 | Bioquímica en suero: colesterol: 4,8 mmol/lβ-colestanol: 122,6 μmol/l | EEG: discreta disfunción neuronalRMC: normalEcocardiograma: normalEco-doppler de troncos supraaórticos: normal | AQDC 750 mg/díaAtorvastatina 40 mg/día |
Valores de referencia: colesterol < 6,1 mmol/l; β-colestanol < 12 μmol/l; 23,25 pentol < 5 mg/mol creatinina; 24,25 pentol < 23 mg/mol creatinina. AQDC: ácido quenodesoxicólico; creat.: creatinina; EEG: electroencefalograma; M: mujer; RMC: resonancia magnética cerebral; V: varón; XCT: xantomatosis cerebrotendinosa; XT: xantomas tendinosos.
Los primeros síntomas de la enfermedad aparecieron durante la primera o segunda décadas de la vida, siendo en todos los casos de carácter neurológico (retraso mental).
Además, dos de ellos fueron intervenidos de cataratas durante la infancia y uno presentó también un cuadro de diarrea crónica.
Las lesiones cutáneas se iniciaron progresivamente 15 años después de la aparición de la sintomatología neurológica, en la segunda o tercera década. Estas lesiones eran unos tumores subcutáneos, firmes, redondos y de características no inflamatorias, localizados sobre las articulaciones, afectando a los tendones aquíleos, extensores de los dedos, rotulianos y del tríceps (figs. 1 y 2). Además, algunos de ellos presentaban xantelasmas. El estudio histológico de estas tumoraciones objetivaba una proliferación de células xantomatosas, sin atipias ni mitosis, abundantes células gigantes multinucleadas tipo Touton y un leve componente inflamatorio intersticial acompañado de colágeno dérmico con degeneración hialina. En algunos casos podían observarse unas hendiduras biconvexas que eran el negativo de los cristales de colesterol disueltos durante el procesamiento de la biopsia. Todo ello era compatible con el diagnóstico de xantoma tendinoso (fig. 3). El examen neurológico de los pacientes mostraba desde retraso mental grave aislado hasta marcha atáxica, amiotrofia de las extremidades e hiperreflexia generalizada, epilepsia o alteraciones psiquiátricas como trastornos del comportamiento e intentos de autolisis.
.")
.")
 (paciente 2). Células xantomizadas separadas por bandas de colágeno.")
Con la sospecha de XCT se realizó la determinación de β-colestanol en suero (pacientes 2, 3 y 4) y de alcoholes biliares en orina (paciente 1). Tras confirmarse el diagnóstico se inició tratamiento con 750 mg/día de AQDC asociado a un inhibidor de la 3-hidroxi-3-metilglutarilcoenzima A (HMG-CoA) reductasa. Las alteraciones bioquímicas se normalizaron rápidamente y se mantuvieron en el rango de los valores controles durante el seguimiento; sin embargo, la sintomatología neurológica permaneció prácticamente igual, o incluso empeoró en alguno de los pacientes. El paciente 3 falleció a los 4 años del diagnóstico por deterioro neurológico progresivo y la paciente 1 fue ingresada en una residencia geriátrica, por lo que la perdimos en el seguimiento.
DiscusiónLa XCT es una enfermedad hereditaria autosómica recesiva incluida en el grupo de las xantomatosis normolipémicas2,3. Nuestros 4 casos se añaden a los otros 4 casos publicados previamente en español, y muestran que la XCT es una enfermedad excepcional o posiblemente infradiagnosticada en nuestro país7–9.
Los pacientes afectos presentan un déficit congénito de la enzima 27α-esterol-hidroxilasa, una enzima clave en la degradación del colesterol. Ello provoca el depósito de sus metabolitos (β-colestanol y alcoholes biliares) en los tejidos, como consecuencia de la falta del efecto de retroalimentación inhibidor del AQDC10. El gen responsable (CYP21A1) se localiza en el brazo largo del cromosoma 2, donde se han identificado más de 50 mutaciones diferentes11,12. La mayoría de casos presenta una sustitución de un aminoácido que comporta la ausencia o una forma inactiva de la enzima5.
Los primeros síntomas suelen aparecer en la primera década de la vida, a menudo en forma de cataratas12,13. Sin embargo, la mayoría de los pacientes no es diagnosticado hasta que no se ha instaurado un cuadro clínico completo14. La diarrea crónica de difícil manejo e inicio en la infancia es también un síntoma infrecuentemente atribuido a la XCT, pero para algunos autores podría tratarse de un síntoma clave para el diagnóstico13. La acumulación de β-colestanol en el cerebro y en el líquido cefalorraquídeo (LCR) es la responsable de la disfunción neurológica. Dado que muchos pacientes presentan atrofia cerebral, se postula que la concentración elevada de β-colestanol en el LCR induciría una apoptosis neuronal2,12.
Los xantomas tendinosos, presentes en el 90–95 % de los casos según las series, son la manifestación cutánea característica13,14. Aparecen durante la segunda o tercera décadas de la vida en forma de nódulos subcutáneos localizados típicamente sobre los tendones aquíleos, pudiendo estar presentes sobre los rotulianos, extensores de los dedos y del tríceps12. La imagen histológica muestra un agregado de células espumosas separadas por bandas de colágeno, sin atipias ni mitosis2,6,10. Como ocurrió en nuestros pacientes, el reconocimiento de los xantomas tendinosos es fundamental para orientar el diagnóstico de XCT en aquellos casos con alteraciones neurológicas, cataratas o diarrea crónica sin diagnóstico.
La XCT se caracteriza también por una alta incidencia de enfermedad coronaria y aneurismas secundarios a una aterosclerosis prematura. Por este motivo se recomienda investigar la presencia de enfermedad cardiovascular12. La muerte ocurre entre la cuarta y la quinta décadas de la vida como resultado del deterioro neurológico progresivo, una parálisis pseudobulbar o un infarto agudo de miocardio13.
La realización de pruebas bioquímicas en suero que incluyan la cuantificación del β-colestanol, paralelamente a un perfil alterado de esteroles característico de la XCT, confirma el diagnóstico, así como la determinación de alcoholes biliares en orina mediante cromatografía de gases-espectrometría de masas10. El estudio genético de las familias permite realizar un diagnóstico temprano en homocigotos pre-sintomáticos y detectar los heterocigotos5,15.
La base del tratamiento es el suministro de los ácidos biliares deficitarios. El AQDC se utiliza como la terapia estándar1. Parece que su uso prolongado podría llegar a detener o, menos probablemente, hacer involucionar la enfermedad, sin la presencia de efectos adversos relevantes12. Hay estudios recientes que proponen el uso asociado de inhibidores de la HMG-CoA reductasa (atorvastatina o simvastatina) ya que, al parecer, su uso combinado reduce las concentraciones de colesterol que se incrementan con la administración de AQDC16–19. Una vez que se instaura el daño neurológico, no se recupera. Para que el tratamiento sea efectivo debería iniciarse ante los primeros síntomas (cataratas o diarreas y alteraciones neurológicas leves); habitualmente cuando han aparecido los xantomas es demasiado tarde para obtener unos resultados satisfactorios. En nuestros casos, el tratamiento se inició tras el desarrollo de los xantomas tendinosos, por lo que no se consiguió la remisión de la enfermedad, pero sí, en algún caso, el enlentecimiento de la progresión.
ConclusiónLa XCT es una enfermedad cuyas claves diagnósticas son la presencia de síntomas neurológicos, cataratas o diarrea crónica en pacientes jóvenes, y xantomas a partir de la segunda década de la vida. Esta enfermedad tiene un tratamiento específico cuya efectividad es mayor cuanto más tempranamente se instaura, y que puede mejorar en algunos casos el grave pronóstico que comporta.
AgradecimientosAgradecemos al Dr. Jordi del Río del Servicio de Neurología, y al Dr. Albert Selva del Servicio de Medicina Interna, por su ayuda en el manejo y seguimiento de los pacientes, y a la Dra. M.a Josep Coll por su ayuda en el diagnóstico biquímico de los mismos.
Conflicto de interesesDeclaramos no tener ningún conflicto de intereses.


