



In January 2012, vismodegib (Erivedge, manufactured by Genentech) became the first selective inhibitor of the Hedgehog signaling pathway to be approved by the US Food and Drug Administration for the treatment of locally advanced and metastatic basal cell carcinoma. The drug selectively binds to Smoothened, a 7-helix transmembrane receptor, thereby inhibiting activation of transcription factors of the glioma-associated oncogene family and suppressing tumor proliferation and growth. Studies published to date have assessed the efficacy of vismodegib according to clinical and radiologic outcomes but little information is available on the molecular mechanisms underpinning the proven clinical efficacy of the drug. This review will cover recent data on the Hedgehog signaling pathway and data from clinical trials with vismodegib in the treatment of basal cell carcinoma, and will consider its use in other types of tumor.
Vismodegib es el primer inhibidor selectivo de la vía de señalización Hedgehog que ha sido aprobado por la Food and Drug Administration (FDA) en EE. UU. para el tratamiento del carcinoma basocelular localmente avanzado y metastásico (Erivedge, Genentech, enero de 2012). Este fármaco se une e inactiva específicamente el receptor de transmembrana-7 Smoothened (SMO), frenando la activación de la familia de factores de transcripción del oncogén asociado a glioma (GLI), suprimiendo la proliferación y el crecimiento tumoral. Los estudios publicados hasta la fecha evalúan la eficacia de vismodegib basándose en criterios clínicos y radiológicos, pero disponemos de escasa información respecto a las bases moleculares que justifican la eficacia clínica probada del fármaco. Esta revisión intenta proporcionar datos actuales respecto a la vía de señalización Hedgehog, datos sobre los ensayos clínicos realizados con vismodegib en el tratamiento del carcinoma basocelular y su uso en otro tipo de tumores.
Basal cell carcinoma (BCC) is the most common skin cancer with an estimated annual incidence of 0.1% to 0.5%.1 It accounts for 80% of nonmelanoma skin cancers.
Several therapeutic alternatives are available, including electrocoagulation, curettage, cryotherapy, topical imiquimod, photodynamic therapy, surgery, and radiotherapy, according to type and site of the tumor and the characteristics of the patient. In some cases however, BCC is particularly aggressive and metastasis may result.2 To date, the therapeutic recommendations for unresectable or metastatic tumors and those inaccessible to radiotherapy have included chemotherapy with platinum, 5-fluoroacyl, vincristine, etoposide, bleomycin, methotrexate, cyclophosphamide, or doxorubicin, either as single agents or in combination. If chemotherapy was contraindicated due to associated comorbidities or adverse effects, palliative or supportive therapy was to be applied. Although Pfeiffer et al.3 claimed that cisplatinum was the most effective chemotherapeutic agent, other authors have reported equally or more effective therapeutic options such as the combination of vincristine, bleomycin, and prednisolone.
In the near future, another option for control of locally advanced tumors not amenable to surgery or radiotherapy may be vismodegib, an inhibitor of the Hedgehog (Hh) signaling pathway.
Hedgehog Signaling PathwayThe first articles were published on the Hh pathway when the Patched (PTCH) receptor was identified during embryonic study of the fruit fly Drosophila melanogaster.4 The Hh pathway appears to be crucial for normal embryonic development of Drosophila, conditioning segmentary polarity and normal morphologic structure.
Several years earlier, Binns et al.5 reported an epidemic of cyclopia in sheep that consisted of the development of congenital defects characterized by the absence of midline facial structures and failure of the prosencephalon to develop into 2 hemispheres (holoprosencephaly). The cause of this malformation was the intake during gestation of a plant of the lily family called Veratrum californicum, which contains an alkaloid steroid called cyclopamine.6,7 Years later, cyclopamine was shown to have a direct role as an inhibitor of the Hh pathway by directly blocking one of the components of this pathway called Smoothened (SMO).8 However, although this agent was very important for study of preclinical models of the Hh signaling pathway, its poor oral bioavailability and acid sensitivity required the development of other synthetic and semisynthetic derivatives that could be administered orally.9
The Hh signaling pathway plays a crucial role in organogenesis during embryogenesis in many species, but in adults it is only active in hair follicles and stem cells, where it plays a major role in maintaining tissue homeostasis and cell repair.10 The mechanism for processing, secretion, and signaling of Hh proteins has been more or less conserved in the phylogenetic scale, although with some differences. Drosophila has a single Hh gene, whereas vertebrates have 3 homologous Hh ligands: Sonic (Sh), Desert (Dh), and Indian (Ih), with the first of these being the most well-known. Dh is implicated in the development of male germ cells and Ih is an important regulator of bone growth and cartilage development.11 Sh is implicated in several activities such as establishing left-right symmetry and development of the central nervous system, eyes, and muscles. The receptor system of these proteins is the Patched 1 (PTCH1) transmembrane protein receptor and a transmembrane SMO protein, which belongs to the G protein coupled receptor family and acts as a mandatory signal transducer for the Hh pathway (Fig. 1). In the absence of a ligand, the PTCH1 receptor suppresses SMO activity; if it binds the ligand, SMO suppression is abolished resulting in modulation and activation of the GLI transcription factors that translocate to the nucleus with induction of transcription of genes such as GLI1,12 the main marker for the Hh signaling pathway. SMO activation requires 2 stages for activation in presence of Hh: phosphorylation of the C-terminal end by the AMPc-dependent protein kinase (PKA), casein kinase 1 (CK1), casein kinase 2 (CK2), and the G protein coupled receptor kinase 2 (GRK2), and their subsequent translocation to the cell membrane through cytoplasmic vesicles. In absence of Hh, several phosphatases can maintain low levels of SMO phosphorylation.
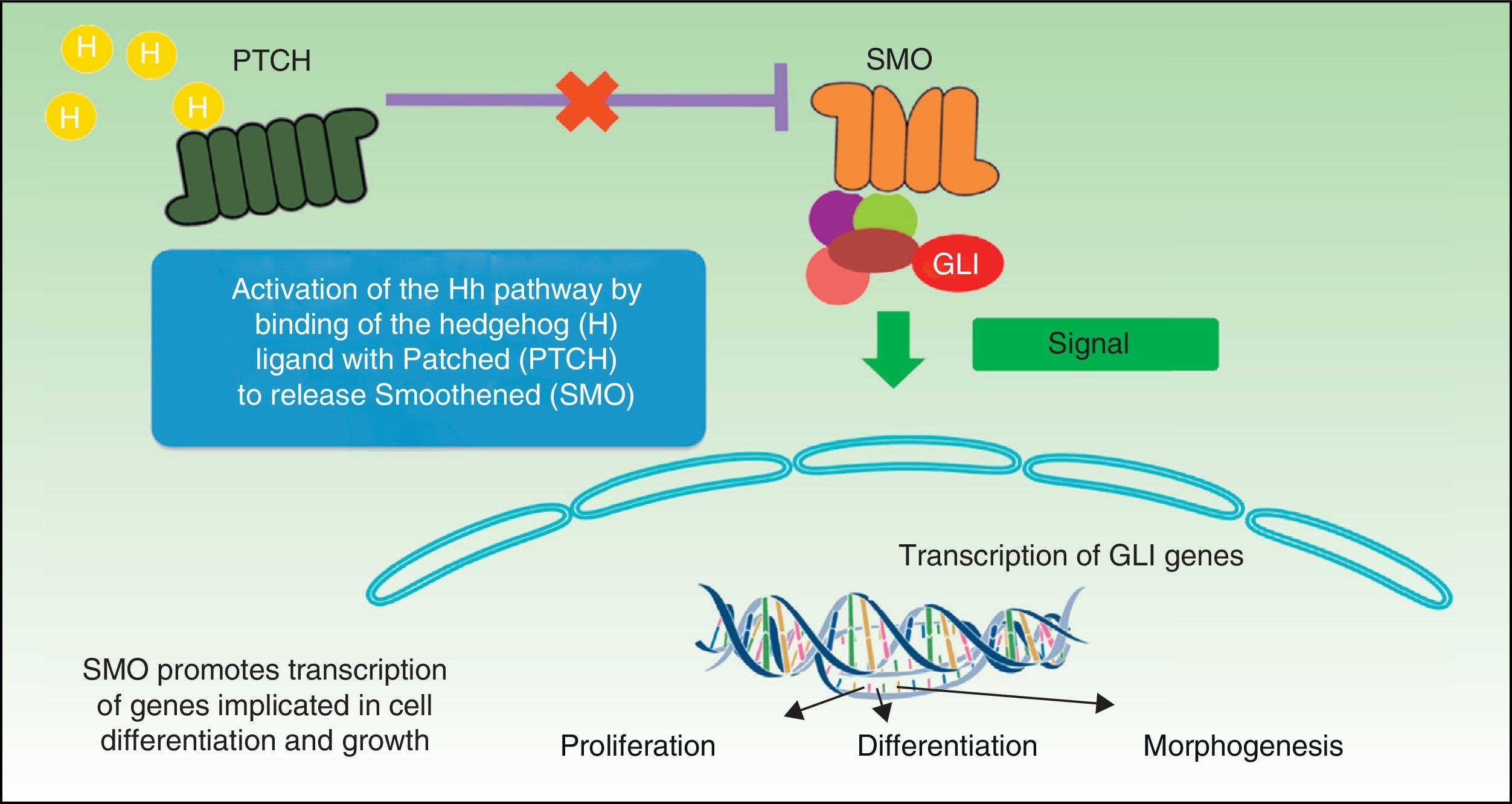
Two signaling modes operate within the Hh pathway, the canonic and noncanonic pathways. The canonic Hh pathway regulates the activation of members of the GLI family of transcription factors. The noncanonic pathway is activated independently of the GLI proteins and in turn is subdivided into 2 signaling pathways, those that do not require SMO (type 1) and those that in addition to not requiring SMO also do not require GLI (type 2). In the canonic Hh signaling pathway, in absence of ligand, PTCH prevents activation of SMO and translocation to the membrane, whereas binding of Hh to the PTCH receptor results in internalization of the ligand-receptor complex and activation by phosphorylation and translocation of SMO. In vertebrates, signaling occurs in the primary cilia, regulating the processing and activation of the GLI protein. The primary cilia are immobile cilia that are present in most vertebrate cells and that comprise a protrusion from the membrane in the apical region of polarized cells. PTCH1 is present in abundance in primary cilia in absence of Hh and is eliminated when Hh is present.13 The fundamental role of the canonic Hh pathway is induction of cell proliferation through induction of genes that encode cyclin D1 and N-myc.14 The noncanonical pathway of type 1 PTCH1 regulates cell survival, such that overexpression of PTCH1 induces apoptosis, as has been observed in several in vitro and ex vivo assays.15 In vertebrates, the 3 Hh ligands have an antiapoptotic effect on culture cells, and this effect is not blocked by SMO antagonists. In this signaling pathway, regulation of the cell cycle is achieved through modulation of the subcellular localization of cyclin B1. Binding of Sh to PTCH prevents PTCH from interacting with cyclin B1, giving rise to an increase in cell proliferation and cell survival.13 In the type 2 canonic SMO-dependent signaling pathway, SMO regulates cytoskeletal actin through GTPases, inducing migration processes, tubulogenesis, and dendritic spine and axon formation in neurones.13
As mentioned above, the Hh pathway is normally inactive during adult life and only remains functional in stem cells, skin, and hair follicles. In vitro and in vivo studies have shown how aberrant reactivation of the Hh pathway can activate genes that promote cell proliferation, giving rise to several types of tumors such as medulloblastomas; rabdomyosarcoma; melanoma; pancreatic, breast, lung, liver, and stomach cancer; and BCC.16,17 Therefore, inhibition of the GLI transcription factors has been proposed as an emerging therapeutic approach with promise in several types of tumors.
There are basically 2 mechanisms by which an increase in the activation of the Hh pathway occurs. In one of these, there is an increase in the expression of Hh bound proteins, which directly leads to increased signaling. In the other, genetic changes in PTCH1 and SMO give rise to constitutively active receptors that also lead to an increase in signal transduction by this pathway.
Molecular Alterations in Basal Cell CarcinomaThe Hh signaling pathway is constitutively activated in BCC.18,19 Mutations have been found in the proteins that regulate the Hh pathway independently of the presence of the Hh ligand, whereas in other human tumors such as ovarian or colorectal cancer, carcinogenesis is activated by an increase in the expression of the Hh ligand in tumor cells, which in turn act on stromal cells through paracrine signaling mechanisms. Mutations in the PTCH1 gene in BCC were identified by studying patients with basal cell nevus syndrome (BNS) or Gorlin syndrome, a hereditary disease in which patients present with multiple BCCs and developmental disorders. PTCH1 mutations have been identified in most of the exons of the gene in patients with BNS and sporadic BCC. In the study conducted by Aszterbaum et al.,20 23 exons from 86 samples of DNA from patients with BNS, 26 from patients with sporadic BCC, and 7 from patients with BNS-associated BCC were analyzed. Of the 26 sporadic BCC analyzed, 11 showed loss of heterozygosity in one or more of the polymorphism markers studied. This suggests that PTCH1 acts as a classic suppressor gene, requiring mutation in 2 alleles to activate tumorigenesis.
In the case of acquired BCC, these mutations in both alleles arise due to postnatal insults, such as exposure to UV light, radiotherapy, chemical carcinogenesis, or random genetic mutations. In contrast, patients with BNS have a greater susceptibility to tumorigenesis, as one of the mutations is inherited through the germ line and so loss of heterozygosity occurs after only one acquired mutation. PTCH mutations were detected in 3 tumors, each one in a different exon of the gene. The mutation led to amino acid substitution in one case, the appearance of a stop codon in another, and a frameshift in the third. These mutations are not usually those associated with exposure to sunlight or induced by UV.
In the case of patients with BNS, mutations were identified in 8 exons in 13 of the 86 patients. The mutations most frequently found corresponded to frameshifts, resulting in gene products with premature chain termination.
Smoothened MutationsAccording to the model proposed for inhibition of SMO by PTCH in the presence of Sh, SMO can only be in its active form in the absence of Sh. In contrast, in patients with sporadic BCC, investigators have identified activating missense somatic mutations in the SMO gene which cause SMO to act as an oncogene. In order to investigate the oncogenic potential of SMO, Xie et al.19 found that overexpression of mutated SMO in the epidermis of transgenic mice gave rise to the formation of epidermal disorders similar those in BCC, with a similar phenotype to those obtained in transgenic mice that express wild type Sh.21
Mutations in the p53 GeneIn addition to PTCH mutations, p53 mutations are common in BCC. Ling et al.22 studied microdissected samples from tumor and adjacent healthy skin by polymerase chain reaction and sequencing. They found allele loss from the PTCH locus in 6 of 8 samples from sporadic BCC and 17 of 19 samples from hereditary BCC, whereas p53 mutations were found in all cases of sporadic disease and only in 7 of 20 hereditary cases. In the cases of hereditary BCC, p53 mutations included single nucleotide deletions and unusual double-base substitutions compared with the pattern of missense mutations induced by UV light in sporadic cases. The high frequency of both mutations and their coexistence suggests that genetic mutations in the PTCH and p53 genes are important for the development of BCC.
Molecular Expression Pattern in Basal Cell CarcinomaIn the analysis of the genetic expression profile of BCC performed by Bonifas et al.,23 BCC expresses high levels PTCH1, GLI1, HIP, WNT2B, and WNT5a mRNA and low levels of c-Myc, c-fos, and WNT4 mRNA compared to normal skin. These alterations suggest that mutations in the Hh pathway play a fundamental role in BCC carcinogenesis. PTCH1, GLI1, and HIP appear to be the most extensively expressed in this type of tumor. The HIP gene encodes a protein able to bind to Sh with an avidity similar to that of PTCH1, such that high levels of mRNA in BCC indicate that the function of this protein could be essential for Hh signaling in humans.
Clinical Trials With VismodegibVismodegib (GDC-0449) was the first drug approved by the Food and Drug Administration (FDA) for the treatment of locally advanced BCC ineligible for surgery or radiotherapy and for metastatic BCC (Erivedge, Genentech, January 2012). This molecule is a 2-aryl pyridine that inhibits the Hh pathway by blocking activation of SMO.3
To date, 1 phase I and 2 phase II trials have been published of vismodegib in patients with advanced and/or metastatic BCC (Table 1).
Summary of Clinical Trials with Vismodegib.
| Authors | Clinical Phase | N Patients With BCC | Response | Response Rate | |||||
| LA | M | CR | PR | SD | P | LA | M | ||
| Von Hoff et al.24 | I | 15 | 18 | 2 (LA) | 9 (M)+7 (LA) | 7 (M)+4 (LA) | 2 (M)+2 (LA) | 60% | 50% |
| Sekulic et al.25 | II | 71 | 33 | 13 (LA) | 14 (LA)+10 (M) | 43% | 30.3% | ||
| Tang et al.26 | II | 42 patients with Gorlin syndrome | Reduction in no. BCC (2 vs 29) and less surgery (0.31 vs 4.4) | ||||||
Abbreviations: CR, complete response; LA, locally advanced; M, metastasis; P, tumor progression; PR, partial response; SD, stable disease.
Von Hoff et al.24 conducted a 2-stage phase I trial that assessed the safety and tolerability of vismodegib in 68 patients, 33 of whom had advanced (n=15) or metastatic (n=18) BCC. In the first stage, which aimed to determine the maximum tolerated dose, 20 patients were evaluated at 3 different doses. They received the drug on Day 1, then had a washout period of 6 days before receiving the same dose from Day 8. Seven patients received 150 mg/d, 9 received 270 mg/d, and 4 received 540 mg/d. No dose-limiting toxicities were reported. Three of the 20 patients had BCC and each received 3 different doses of vismodegib (150, 270, and 540mg). In stage 2, the aim was to obtain additional information on pharmacokinetics, pharmacodynamics, and safety. A further 48 patients were recruited, 30 of whom had BCC: 16 patients received 150mg/d and 14 received 270mg/d. Follow-up was every 4 weeks and involved a physical examination, electrocardiogram, biochemistry, and hematology. For those patients with radiologically assessable disease (above all in metastatic BCC), the tumor assessment was based on response evaluation criteria in solid tumors (RECIST) (version 1.0). For patients with radiologically nonassessable disease, follow-up involved physical examination, such that complete response was considered as disappearance of the palpable or visible tumor, and a partial response as a reduction of more than 50% in the palpable or visible tumor diameter. Of the 18 patients with metastatic BCC, 9 achieved partial response, 7 had stable disease, and 2 had tumor progression. The overall response rate for patients with metastatic BCC was 50%. Of the 15 patients with locally advanced BCC, 2 achieved complete response, 7 achieved partial response, 4 had stable disease, and 2 had tumor progression. The overall response rate in this group of patients was 60%. The most frequently observed adverse effects, all grade 2 to 3, were muscle cramps, dysgeusia, anorexia, and weight loss. The dose established as the most appropriate was 150mg/d, as no increases were observed in plasma concentration in parallel with and proportional to increases in the orally administered dose. Vismodegib undergoes minimal metabolism and more than 98% of the drug is eliminated unchanged by the biliary route (82% is recovered in feces and 4.4% in urine). Results from in vivo studies suggest that the drug acts as an inhibitor of the CYP2C8, CYP2C9, and CYP2C19 cytochromes and BRCP (breast cancer resistance protein). The adverse effects may be increased if the drug is administered concomitantly with other drugs that inhibit P-glycoprotein, such as clarithromycin, erythromycin, and azithromycin. The estimated half-life of vismodegib is 12 days after a single dose and 4 days after continuous daily administration.
Phase II Clinical TrialsSekulic et al.25 conducted a phase II clinical trial that formed the basis for approval by the FDA. They enrolled 104 patients with metastatic BCC (n=33) and locally advanced BCC (n=71). Of these, 8 were excluded from the trial because the pathologist could not detect BCC in the initial biopsy. All patients received vismodegib at a dose of 150mg/d until tumor progression was observed or severe pharmacologic toxicity occurred. The primary efficacy outcome measure was response rate. For patients with metastatic BCC, the RECIST guidelines were used (version 1.07) whereas for patients with locally advanced BCC, response was defined as a decrease of 30% or more in tumor size or complete disappearance. Progressive disease was defined as an increase of 20% or more in the size of the visible tumor or radiologically measured tumor, the appearance of a new ulceration, or new lesions. For patients with multiple lesions, the sum of the largest diameters was used to determine response and progression. In the group with metastatic BCC (n=33), 10 patients attained partial responses (30.3%) and there were no complete responses. The mean duration of objective response in this group of patients was 7.6 months. In the group with locally advanced BCC (n=63), 13 patients attained complete response (20.6%) and 14 had partial responses (22.2%). The overall response rate in this group of patients was 43%. The mean duration of response was 7.6 months. In addition, in 34 of the 63 patients (54%), no residual BCC was observed in subsequent biopsies. The most frequently observed adverse effects, all grade 2 or 3, were muscle cramps, weight loss, fatigue, and loss of appetite. The most serious adverse effects were observed in 7 patients (one with metastatic BCC and the rest with locally advanced BCC): 3 deaths of unknown cause, myocardial infarction, meningeal disease, and cerebrovascular accident. The relationship between deaths and the drug is not known at present. At a molecular level, increased expression of the genes that code for factor GLI1 and patched homologous receptor 2 (PTCH2) genes was noted and confirmed in skin tumor biopsies prior to treatment with vismodegib, compared with biopsies of healthy skin from other subjects.
The other phase II clinical trial was published by Tang et al.26 This study was a randomized, double-blind, placebo-controlled study that aimed to investigate the efficacy of vismodegib in patients with Gorlin syndrome. The study enrolled 42 patients with this syndrome who were randomized 2:1 to receive oral vismodegib at a dose of 150mg/d (n=26) or placebo (n=15) for a period of 18 months. The primary efficacy outcome measure was the comparative rate of new BCC eligible for surgical resection (those with a diameter ≥3mm on the nose or periorbital area, ≥5mm on any part of the face, or ≥9mm on the trunk and thighs). The secondary efficacy outcome measures were decrease in the rate of appearance of small BCC (≤5mm) on the upper third of the back, decreased size of surgery-eligible BCC, effect duration on BCC after discontinuation of the drug, and changes in expression of the Hh gen in BCC. Clinical follow-up was performed monthly for the first 9 months and then every 3 months for the remaining 9 months. Only 38 patients completed the scheduled study visits in the first 3 months. In addition, molecular studies were undertaken to assess the degree of inhibition of the Hh pathway through analysis of levels of mRNA encoding GLI1 in skin biopsies performed at baseline and within 1 month of initiating vismodegib. A decrease was observed in the appearance of new BCC eligible for surgery in the vismodegib group compared to placebo (2 vs 29 new BCC per year, P<.001). Vismodegib also reduced the size of existing tumors, expressed as a percentage of change in the sum of largest diameters (–65% vs –11%, P=.003). Patients in the vismodegib group underwent fewer surgical procedures compared to the placebo group (mean number of procedures per patient 0.31 vs 4.4 for placebo, P<.001). Almost all tumors responded to vismodegib, with almost complete clinical remission in some patients. In skin biopsy and molecular studies, among the patients who received vismodegib for the first 3 months, 46% of the biopsies showed presence of residual tumor, and among the lesions that appeared clinically resolved, presence of residual tumor was seen in only 17%. At a molecular level, the effect of vismodegib was also associated with a decrease in Hh signaling, with a decrease of up to 90% in expression of GLI1 mRNA in biopsies performed after 1 month of treatment and a reduction in tumor proliferation, assessed through expression of Ki67. However, the authors reported no changes in cell apoptosis, assessed by measurement of caspase 3. The most frequently observed adverse effects, all grade 1 to 3, were dysgeusia, muscle cramps, alopecia, and weight loss. No serious adverse effects were reported. After 8 months of treatment, 7 of the 26 patients (27%) who received the drug discontinued therapy due to adverse effects. These resolved between 1 and 3 months later.
Vismodegib in Special SituationsVismodegib and GestationVismodegib is considered a category D drug by the FDA. In studies with rats, teratogenicity was observed at 20% of the daily recommended dose for humans. Effects included the appearance of craniofacial defects, open perineum, and digital fusions.27
Vismodegib in ChildrenThe efficacy and safety of vismodegib in human children have not been studied. In animal studies, different abnormalities were observed at doses of 20% to 40% of the recommended adult dose, with the appearance of early epiphyseal closure and dental abnormalties.27
Vismodegib in Elderly PatientsTo date, not enough patients aged ≥65 years have been included in clinical trials to assess whether differences in terms of pharmacokinetics, safety, and efficacy are present in elderly patients. Because most metastatic and/or locally advanced BCCs occur in eldery patients, more postmarketing studies should be performed to provide data in these patients.28
Mechanisms of Resistance to VismodegibThe topic of acquired resistance to drugs is particularly pertinent in the case of kinase inhibitors. Soon after the recent introduction of vismodegib as an inhibitor of the Hh signaling pathway, the first report of resistance to the therapeutic group targeting the SMO receptor was published.29 The concept has been extended to cover the phenomenon of acquired resistance to drugs targeting molecules related to the G-protein coupled receptor. Rudin et al.30 published the first case of medulloblastoma treated with vismodegib. Good initial response was attained although loss of efficacy occurred after 3 months. The patient was a 26-year-old man with metastatic medulloblastoma who started treatment with vismodegib at a dose of 150mg/d. He achieved a rapid and notable initial tumor regression; molecular analysis of the primary tumor and metastases prior to treatment revealed the existence of a mutation in PTCH1 (PTCH1-W844C), as well as overregulation of different genes implicated in the Hh pathway, thereby confirming the premise that the disease was induced by hyperactivation of this signaling pathway. Subsequently, Yauch et al.31 attempted to elucidate the nature of the resistance mechanism in this patient. They collected a biopsy of the tumor lesion and identified, through genetic sequencing, a heterozygous missense mutation at position 1497 of SMO corresponding to switching aspartic acid (Asp) for histidine (His) in codon 473 (D473H). This mutation was not detected in pretreatment biopsies, although it may have been present at levels below the limit of detection. In turn, Dijkgraaf et al.,32 in in vitro studies, found a new mutation in SMO through induction of mutations in regions of SMO important for interaction with vismodegib. Most of the 21 new mutations that showed deficiency for vismodegib binding were inactive in terms of signaling, and were not expressed on the cell surface. However, mutation SMO-E518A showed substantial activity and partial resistance to the presence of 1μmol/L of vismodegib. One way of approaching resistance generated by specific mutations is the development of second-generation inhibitors that conserve activity in the presence of mutations. This approach has been used successfully in the case of resistance to drugs targeting epidermal growth factor receptor (EGFR). An example is nilotinib, a second-generation inhibitor that showed clinical activity in patients with chronic myeloid leukemia resistant to imatinib.33 A valid alternative could be the use of antagonists of the Hh signaling pathway with a different mechanism of action to vismodegib. For example, it has recently been found that itraconazole inhibits the Hh pathway through a mechanism other than the cyclopamine mechanism, although the details have not yet been elucidated.34 The use of antagonists of the Hh pathway with targets other than SMO, such as GANT61, which blocks GLI function, could be considered as a strategy in the context of resistance to vismodegib.35
Use of Vismodegib in Other TumorsHedgehog Signaling Pathway Alteration in Other TumorsAn aberrant Hh signaling pathway can trigger the initiation, proliferation, and propagation of different types of cancer through activation of a ligand-dependent or ligand-independent pathway. In the ligand-independent pathway, genetic mutations in different components of the Hh pathway, such as mutations that lead to loss of PTCH or SUFU function or increased activity of SMO, and missense mutations in GLI1 and GLI3, favor tumor cell survival and growth, as observed in the case of BCC and medulloblastoma. The ligand-dependent pathway may be autocrine if the Hh ligand produced by the tumor cell acts on neighboring cells to stimulate propagation and survival, or paracrine when the Hh ligand secreted by the epithelium stimulates the underlying stroma to propagate proliferation and malignant transformation. Autocrine signaling has been observed in cases of lung cancer, pancreatic cancer, and prostate cancer, whereas the paracrine pathway has been isolated in cases of ovarian and colorectal cancer.36 Moreover, tumorigenic transition is influenced by the stage of the neoplastic process in which the Hh signaling variant intervenes. For example, in cases of BCC and medulloblastoma, alteration of the Hh pathway is important for initiating tumor formation and maintenance. In cases of colon cancer and pancreatic cancer, Hh alteration is important for tumor maintenance, but not for tumor formation. In other types of cancers, such as breast, ovarian, prostate, and lung cancer, it is known that this pathway is implicated, but the exact role has not been elucidated.37 Specifically, in cases of pancreatic, ovarian, and colon cancers, and leukemia, different clinical trials are in progress to assess the efficacy of vismodegib alone or in combination with other chemotherapeutic regimens.
ConclusionIn the last decade, substantial progress has been made in the understanding of the functions and mechanisms of action of Hh protein in the development of cancer. Although not all mechanisms of the Hh signaling pathway have been fully studied, it is evident that an aberrant Hh pathway favors tumor growth and proliferation, making it more aggressive and more likely to metastasize. Inhibition of the Hh pathway is, therefore, a new, promising, and selective approach for treatment of certain types of advanced malignant neoplasms, such as BCC and medulloblastoma. Initial clinical trials performed with the oral SMO antagonist GDC-0449, show good efficacy and safety in BCC and in medulloblastoma. Preliminary studies have laid the foundations for the use of these inhibitors in other types of cancers. In some cases, clinical trials have been published with promising results. However, more experience is needed to truly assess their long-term efficacy, administration time, and the possible appearance of new adverse effects.
Ethical ResponsibilitiesProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed their hospital's protocol on the publication of data concerning patients and that all patients included in the study have received sufficient information and have given their written informed consent to participate in the study.
Right to privacy and informed consentThe authors declare that patient data do not appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Ruiz V, Alegre M, López-Ferrer A, Garcés JR. Vismodegib: revisión. Actas Dermosifiliogr. 2014;105:744–751.


