





La urticaria vasculitis (UV) es un subtipo de vasculitis caracterizada clínicamente por lesiones de tipo urticarial e histológicamente por vasculitis necrotizante.
ObjetivoEl objetivo de este trabajo es el estudio de los pacientes con urticaria vasculitis que han sido atendidos en el Servicio de Dermatología del Hospital Universitario Virgen del Rocío (Sevilla, España) para determinar las características de presentación clínicas e histológicas, así como el despistaje de hipocomplementemia, en estrecha relación con enfermedad sistémica asociada.
Material y métodosSe han extraído de la base de datos de nuestro servicio los casos de urticaria vasculitis confirmados histológicamente durante un período de 10 años.
ResultadosQuince pacientes fueron diagnosticados de urticaria vasculitis (9 mujeres y 6 hombres, con una mediana de edad de 51 años). En 14 pacientes (93%) las lesiones persistían más de 24 horas. En 9 pacientes (60%) las lesiones se resolvían con púrpura e hiperpigmentación residual. Siete pacientes (47%) presentaron niveles disminuidos de complemento en sangre, 12 pacientes (80%) asociaron síntomas extracutáneos y 8 pacientes (53%) asociaban enfermedad sistémica, siendo la entidad más frecuente el lupus eritematoso sistémico.
ConclusionesLa urticaria vasculitis constituye a menudo una entidad infradiagnosticada, con una variable respuesta al tratamiento y en la que la existencia de niveles de complemento disminuidos y de síntomas extracutáneos puede ser indicativa de enfermedad sistémica asociada.
Urticarial vasculitis is a subtype of vasculitis characterized clinically by urticarial lesions and histologically by necrotizing vasculitis.
ObjectiveTo study the clinical and histologic features of urticarial vasculitis in patients seen in the dermatology department of Hospital Universitario Virgen de Rocío in Seville, Spain, and to examine the association between hypocomplementemia and systemic disease.
Material and methodsWe performed a chart review of histologically confirmed cases of urticarial vasculitis in the database of our department covering a period of 10 years.
ResultsFifteen patients (9 women and 6 men with a median age of 51 years) were included. In 14 patients (93%), the lesions persisted for more than 24hours, and in 9 cases (60%) the lesions resolved leaving residual purpura or hyperpigmentation. Seven patients (47%) had low complement levels in the blood, 12 (80%) had extracutaneous symptoms, and 8 (53%) had associated systemic disease, the most common of which was systemic lupus erythematosus.
ConclusionsUrticarial vasculitis may be underdiagnosed. Response to treatment is variable, and hypocomplementemia and extracutaneous symptoms may indicate the presence of associated systemic disease.
La urticaria vasculitis (UV) es una vasculitis de pequeño vaso caracterizada clínicamente por lesiones urticariales que muestran en la histología una vasculitis leucocitoclástica. Puede ser idiopática o estar asociada a enfermedades del tejido conectivo (lupus eritematoso sistémico [LES] y síndrome de Sjögren), neoplasias (linfoma, gammapatía monoclonal), infecciones crónicas por el virus de la hepatitis B y C, virus de Epstein-Barr, Borrelia burgdorferi, enfermedad del suero, fármacos (cimetidina, fluoxetina, procainamida, atenolol, sulfametoxazol, paroxetina, valproato sódico, ciprofloxacino) y exposición solar1. Las formas idiopáticas pueden cursar con niveles normales de complemento o con niveles disminuidos; en estas últimas la sintomatología sistémica es más frecuente. La UV constituye un criterio mayor del síndrome de UV hipocomplementémico (SUVH), desorden autoinmune infrecuente con criterios anatomoclínicos bien definidos y que se manifiesta en el 5% de las urticarias crónicas2.
La UV se da con más frecuencia en los adultos, en la cuarta década de la vida, siendo escasos los casos en la edad pediátrica2. Tiene más prevalencia en las mujeres, constituyendo del 60 al 80% de los casos..
Se postula que la patogénesis de la UV está mediada por un mecanismo de hipersensibilidad tipo 3, en el que se forman complejos inmunes por la generación de autoanticuerpos inmunoglobulina G o de forma exógena en respuesta a fármacos o determinadas infecciones virales. La diana inmunológica de estos autoanticuerpos Ig G se encuentra en la subunidad proteica C1q de factor del complemento C1. De este modo, se forman inmunocomplejos circulantes, presentes en el 30-75% de pacientes con UV, que activan el sistema de complemento por la vía clásica.
Clínicamente, la UV se caracteriza por placas urticariales que pueden ser dolorosas o producir quemazón, duran más de 24 horas y se resuelven con púrpura o hiperpigmentación. Los pacientes pueden asociar angioedema, artralgias, dolor abdominal o torácico, fiebre, enfermedad pulmonar o renal, epiescleritis y uveítis. Las manifestaciones sistémicas se presentan con mayor frecuencia en las formas hipocomplementémicas, y las asociadas a enfermedad sistémica (LES, la enfermedad mixta del tejido conectivo, artritis reumatoide, fiebre mediterránea familiar, artritis crónica juvenil, etc.) o en el contexto del SUVH1,2.
Con este estudio se pretende describir las características clínicas e histológicas de los pacientes con UV que han sido atendidos en el Servicio de Dermatología de nuestro hospital, así como determinar los niveles de complemento en sangre, la existencia de enfermedad sistémica asociada y la terapéutica empleada.
Material y métodosMaterialPacientes con UV atendidos en las consultas del Servicio de Dermatología del Hospital Universitario Virgen del Rocío de Sevilla desde el 1 de enero de 1998 hasta el 31 de diciembre de 2008, considerando como criterio clínico las lesiones urticariales, e histológico los hallazgos de vasculitis leucocitoclástica.
MétodoEstudio retrospectivo y descriptivo de la epidemiología, clínica y opciones terapéuticas de los casos recogidos. Para ello se revisaron las historias clínicas, pruebas de laboratorio y biopsias realizadas. La hipocomplementemia se definió como niveles de C3<90mg/dl (normal: 90-180), C4<10mg/dl (normal: 10-40) y C1q<10mg/dl (normal: 10-25). Las variables incluidas en el estudio fueron: edad, sexo, características clínicas, niveles de complemento en sangre, niveles de anticuerpos antinucleares (ANA), asociación con enfermedad sistémica y tratamiento realizado. Se realizó estadística descriptiva de las variables del estudio y se emplearon frecuencias absolutas y relativas en el caso de las variables cualitativas. Las variables cuantitativas se resumieron mediante p50 (p25-p75) (mediana, rango intercuartílico). Para analizar la asociación entre las variables cualitativas y los 2 grupos en los que se dividieron los pacientes según las cifras de complemento (normocomplemetémicos e hipocomplementémicos) se realizó el test exacto de Fisher, considerándose significativo un valor de p<0,05.
ResultadosSe incluyeron 15 pacientes (60% mujeres y 40% hombres). La mediana de edad al diagnóstico fue de 51 años, con un rango intercuartílico de 23-56. La mediana de duración de la enfermedad fue de 24 meses (12-60). Las características demográficas, los niveles de complemento y la asociación con enfermedad sistémica se han resumido en la tabla 1.
Variables epidemiológicas, niveles de complemento en sangre y enfermedades sistémicas asociadas en pacientes con urticaria vasculitis
| Paciente n° | Edad (años) | Sexo | Complementemia | Enfermedad sistémica asociada |
| 1 | 53 | F | Baja | SUVH |
| 2 | 69 | F | Baja | Artristis reumatoide |
| 3 | 23 | F | Baja | Enfermedad mixta del tejido conectivo |
| 4 | 50 | F | Baja | |
| 5 | 66 | F | Baja | Lupus eritematoso sistémico |
| 6 | 6 | F | Baja | Fiebre mediterránea familiar |
| 7 | 8 | M | Normal | Artritis crónica juvenil |
| 8 | 52 | M | Normal | Lupus eritematoso sistémico |
| 9 | 47 | F | Baja | No |
| 10 | 80 | F | Normal | No |
| 11 | 33 | F | Normal | No |
| 12 | 51 | M | Normal | No |
| 13 | 56 | M | Normal | No |
| 14 | 52 | M | Normal | No |
| 15 | 21 | M | Normal | No |
SUVH: síndrome de urticaria vasculitis hipocomplementémico.
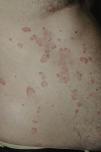
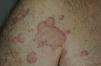
Todos los pacientes presentaban en la exploración clínica placas urticariales de predominio en el tronco y la raíz de los miembros (figs. 1 y 2).
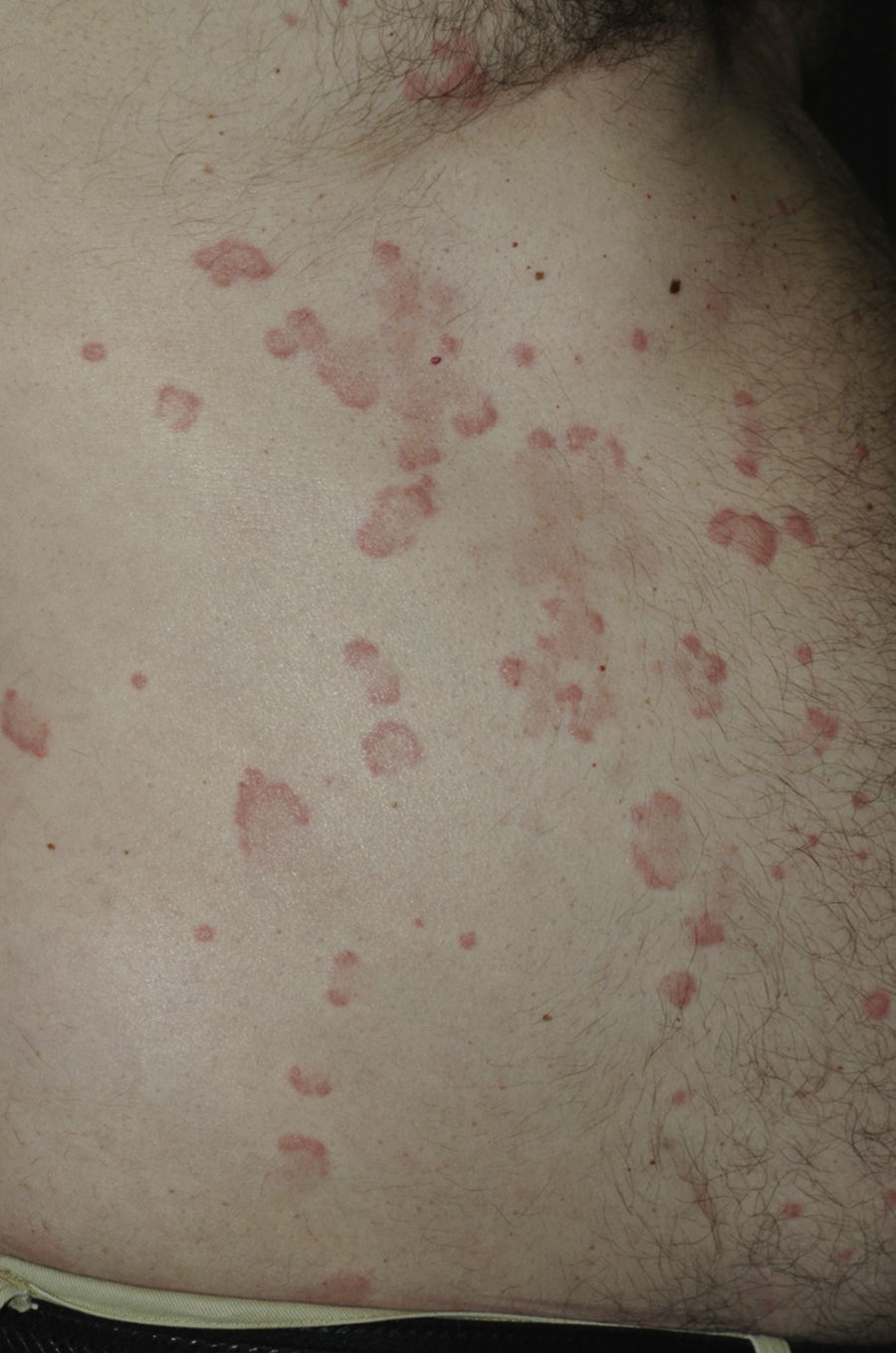
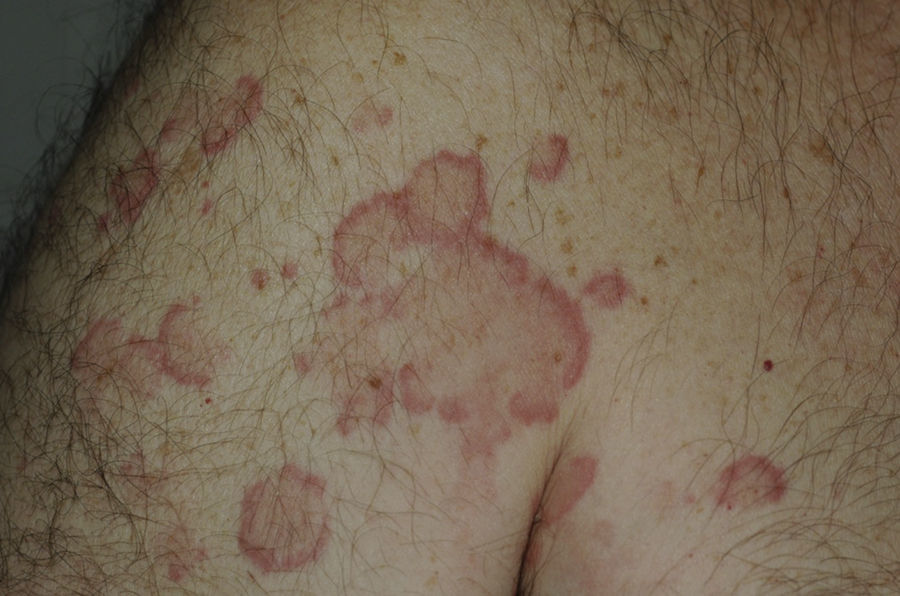
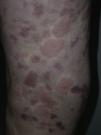
En 14 pacientes (93%) las lesiones persistieron más de 24 horas, siendo del 100% en los pacientes que presentaban hipocomplementemia. Las lesiones producían dolor o sensación de quemazón en 2 pacientes (13%), mientras que en 9 (60%) se resolvían con hiperpigmentación o púrpura residual (fig. 3).
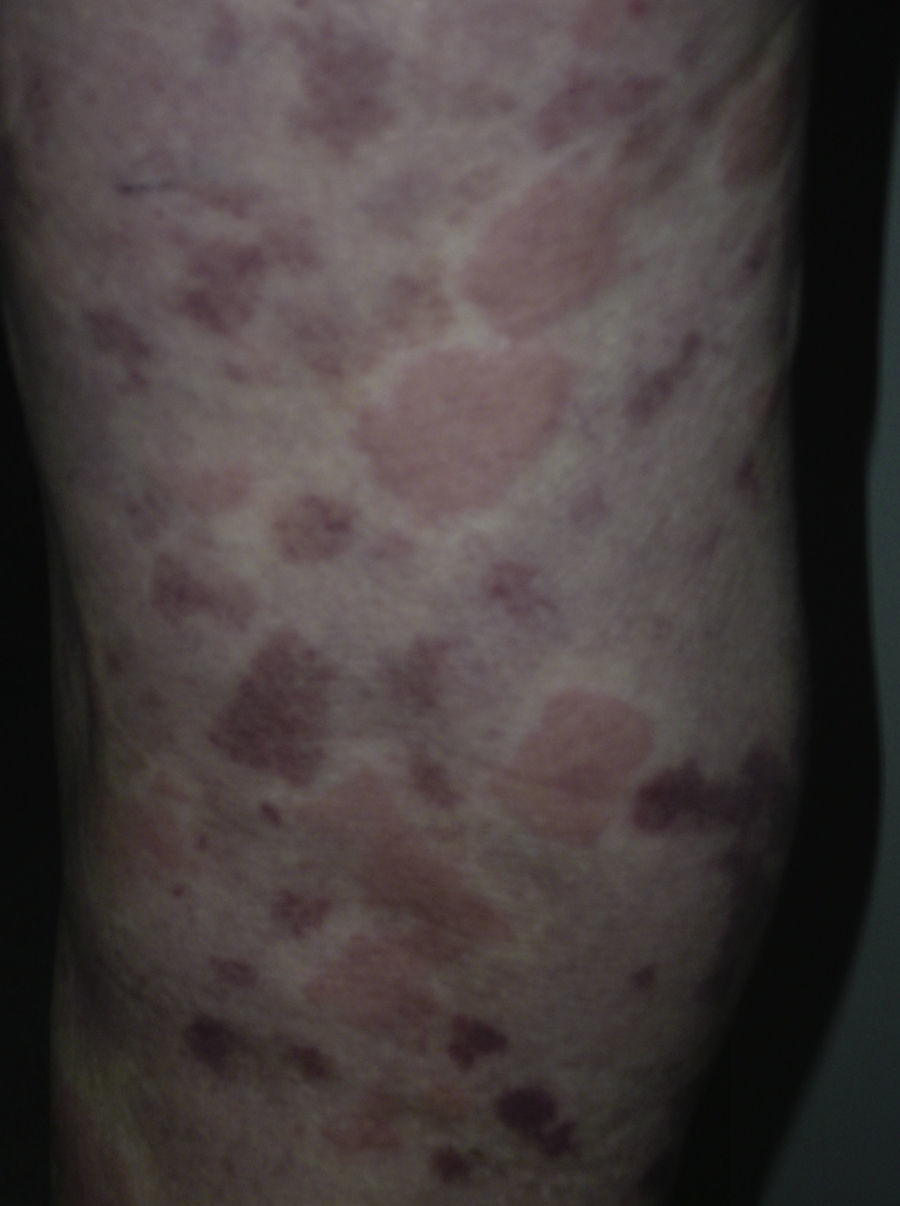
En 2 casos (13%) las lesiones urticariales se acompañaron de angioedema. Uno o más síntomas extracutáneos se encontraron en 12 pacientes (80%), siendo la artralgia y la fiebre los más comunes.
Los pacientes se subdividieron en casos normocomplementémicos e hipocomplementémicos en función de niveles normales o disminuidos de complemento en sangre, respectivamente. De este modo encontramos 8 pacientes (53%) con normocomplementemia y 7 (47%) con hipocomplementemia. De estos, las fracciones C3, C4 y C1q del complemento se mostraron disminuidas en 4 pacientes (57,14%), 2 pacientes (28,57%) presentaban disminución de la fracción C3 y C1q y un paciente (14,28%) tenía disminuida la fracción C4 (tabla 2).
Hallazgos clínicos según los niveles de complemento
| Hallazgos clínicos | Hipocomplementemia | Normocomplementemia | ||
| (n=7) | % | (n=7) | % | |
| Duración lesiones >24h | 7 | 100 | 7 | 87,5 |
| Duración lesiones <24h | - | - | 1 | 12,5 |
| Dolor o quemazón | 2 | 28,6 | - | - |
| Hiperpigmentación residual | 4 | 57,1 | 5 | 62,5 |
| Angioedema de cara o manos | 2 | 28,6 | - | - |
| Artralgia asociada a inicio de enfermedad | 2 | 28,6 | 2 | 25 |
| Enfermedad obstructiva pulmonar | 1 | 14,3 | - | - |
| Dolor abdominal | 1 | 14,3 | - | - |
| Fiebre | 4 | 57,1 | 2 | 25 |
| Depósitos de IG o C3 | 2 | 28,6 | 1 | 12,5 |
| Pared vasos | - | - | - | - |
| Membrana basal | 2 | 28,6 | 1 | 12,5 |
De los 7 pacientes (47%; todas de sexo femenino) que mostraban hipocomplementemia 6 (88%) tenían enfermedad sistémica. Solo aparecieron casos de angioedema en 2 pacientes con hipocomplementemia, en uno con SUVH y en otro con enfermedad mixta del tejido conectivo.
En 5 casos (33%) del total de la muestra se halló positividad para ANA, asociándose en todos estos casos enfermedad sistémica. El 100% de los pacientes con ANA positivos presentaba niveles bajos de complemento, encontrándose por tanto una asociación significativa entre la hipocomplementemia y la presencia de ANA positivos (p=0,002).
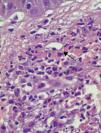
En los 15 casos se halló en el estudio histológico alteraciones compatibles con vasculitis leucocitoclástica (fig. 4).
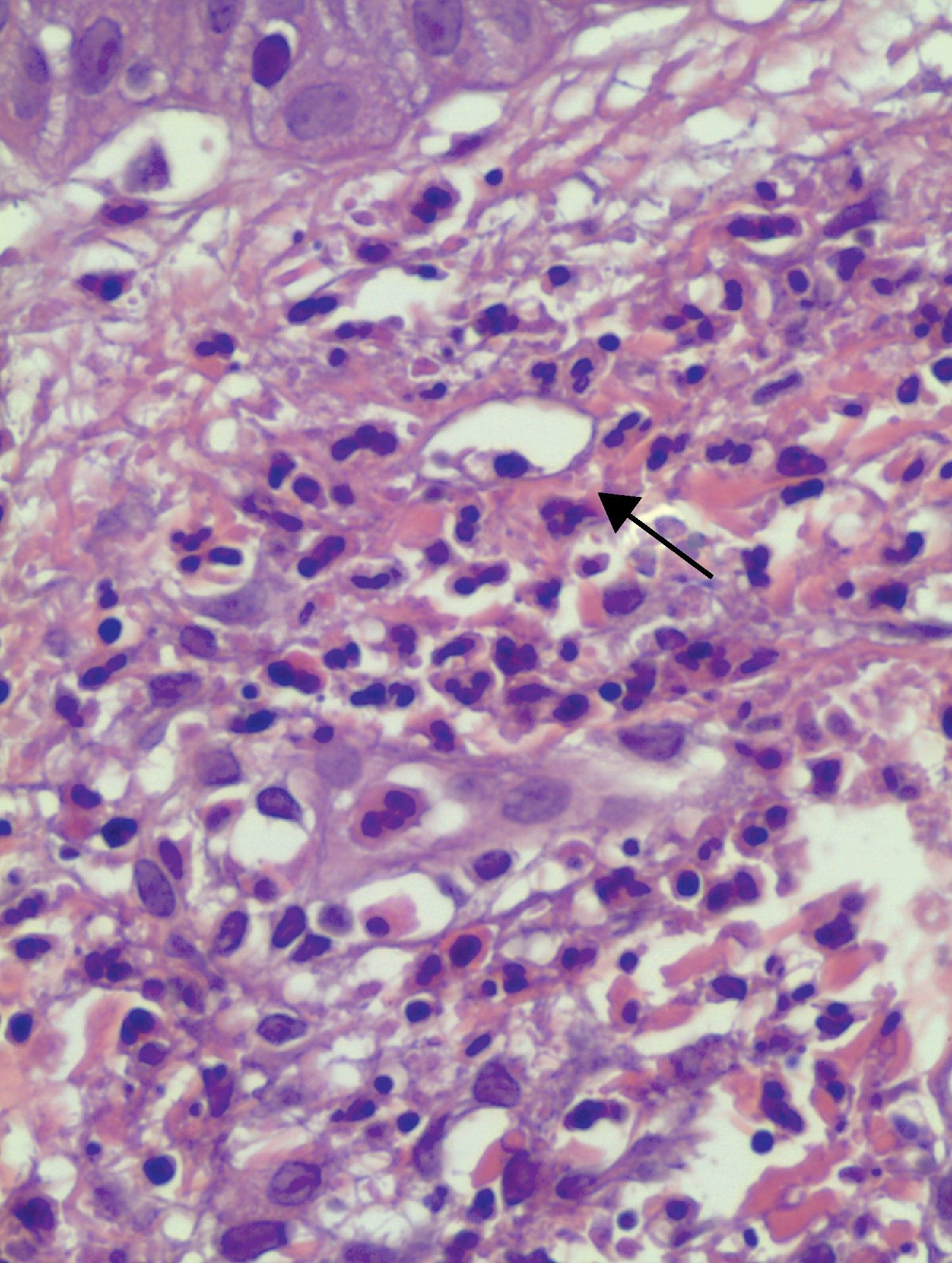
De los 8 pacientes (53%) a los que se realizó inmunofluorescencia directa (IFD), 3 (37,5%) presentaron IFD positiva; 2 mostraban depósito granular de C3 en la unión dermoepidérmica, ambos en el contexto de enfermedades sistémicas (LES y enfermedad mixta del tejido conectivo) y de niveles bajos de complemento en suero; el otro caso positivo presentaba depósito lineal Ig M en la unión dermoepidérmica. El resto de las IFD resultaron negativas.
El modo de presentación de UV fue de forma aislada en 7 de los pacientes (46,66%) y en el contexto de una enfermedad sistémica en 8 pacientes (53%); en 7 de estos coincidió con la enfermedad y en uno la precedió. La enfermedad que se presentó con mayor frecuencia fue el LES en 3 casos (37,5%), y un caso (12,5%) de enfermedad mixta del tejido conectivo, artritis reumatoide, fiebre mediterránea familiar, artritis crónica juvenil y SUVH.
Respecto al tratamiento, en la tabla 3 describimos los fármacos que se utilizaron. Ocho de los 15 casos (53,33%) respondieron a la combinación de antihistamínicos y corticoides orales. En 2 casos (13,33%) se utilizó colchicina con buena respuesta, un paciente (6,66%) respondió a indometacina, otro (6,66%) a dapsona y 3 (20%) estaban siendo tratados con bolos de rituximab y ciclofosfamida por su enfermedad de base (tabla 3).
DiscusiónLa UV es una entidad clinicopatológica de escasa incidencia, quizás por un infradiagnóstico. Comprende un espectro clínico que va desde la afectación cutánea exclusiva a la asociación con enfermedades sistémicas, hecho que se da con mayor frecuencia en las formas hipocomplementémicas. En nuestra experiencia consideramos que los 15 casos registrados representan un número escaso para un período de estudio de 10 años en un área sanitaria de la que dependen 600.000 personas, hecho en consonancia con la consideración de que es una enfermedad infradiagnosticada1–4. A este respecto Tosoni et al.1 manifiestan en su estudio de 47 pacientes con UV la discrepancia ante los criterios clásicos de diagnóstico clínico que podrían conducir a un infradiagnóstico, ya que menos de la mitad de los pacientes presentaban lesiones de más de 24h de duración, menos del 9% refería sensación de quemazón y la púrpura o hiperpigmentación residual fue objetivada solo en un 6% de los pacientes. Por ello proponen que la duración y las características individuales de las lesiones, parámetros no exentos de subjetividad, no deben ser considerados criterios mayores y postulan que deberían ser biopsiadas todas las urticarias crónicas refractarias a antishistamínicos. En nuestra serie, como en la mayoría de estudios previos, podemos observar cómo la práctica totalidad de los pacientes (93%) presentaron lesiones de más de 24h de duración, que se resolvieron con púrpura o pigmentación residual5,6.
En nuestro estudio la asociación con una enfermedad sistémica se produce en más de la mitad de los pacientes, porcentaje muy superior a lo encontrado en otros artículos5,6. Este dato se puede deber en gran medida a que 7 de los 8 pacientes con UV que mostraron asociación con una enfermedad sistémica ya habían sido diagnosticados de la misma en el momento de aparición de las lesiones de UV. Esto pudo condicionar un aumento de la sospecha diagnóstica de UV en estos pacientes con enfermedad sistémica de base y lesiones urticariformes compatibles. En nuestra serie es destacable el caso de fiebre mediterránea familiar con mutación V726A del gen MEFV y UV que se da en una de las pacientes infantiles, no encontrando datos de dicha asociación en la revisión bibliográfica realizada7–9.
Asimismo, la UV puede formar parte del SUVH descrito por McDuffie en 197310, y para el que Schwartz estableció unos criterios y menores11. A diferencia de los casos de UV hipocomplementémica, que puede o no asociar signos extracutáneos, el SUVH ha de presentar necesariamente una serie de manifestaciones extracutáneas para cumplir los criterios establecidos que lo definen. Desde la caracterización del SUVH solo se han publicado 100 casos en adultos, y tan solo 6 casos en niños12,13. En nuestro estudio destaca una paciente con este infrecuente síndrome que cumplía tanto los criterios mayores como los menores del mismo.
La mayor prevalencia del sexo femenino es otro dato coincidente con estudios previos, así como que la UV asociada a hipocomplementemia afecta de manera casi exclusiva a mujeres1,2,5. En nuestro estudio todos los pacientes que mostraron niveles bajos de complemento en suero fueron mujeres.
La edad de presentación suele ser en torno a los 40 o 50 años. De este modo son llamativos los 2 casos de UV encontrados en la edad pediátrica en nuestra serie, asociación muy poco descrita en artículos revisados7,12,13. Ambos casos se hallaban en el contexto de enfermedades sistémicas, uno asociado a fiebre mediterránea familiar y otro a artritis crónica juvenil.
Coincidiendo con los artículos que registran un mayor número de casos, se encontraron uno o más síntomas extracutáneos en el 80% de los pacientes, la fiebre (40%) y la artralgia (27%) fueron los más frecuentes1,5,6. Destaca que los pacientes con hipocomplementemia mostraban mayor tendencia a presentar dolor o quemazón en las lesiones y a asociar sintomatología sistémica como fiebre y artralgias, en comparación con los pacientes con normocomplementemia. Este dato coincide con lo publicado en otras series, en las que se encuentra comparativamente una mayor prevalencia de síntomas extracutáneos en pacientes con niveles disminuidos de complemento1,5,6.
Histológicamente la UV se caracteriza por una vasculitis leucocitoclástica. La biopsia de una lesión activa es la prueba de referencia para el diagnóstico de la UV. La histología de la UV con frecuencia pone de manifiesto una vasculitis leucocitoclástica14,15. En cuanto a los hallazgos en la IFD se pueden objetivar depósitos lineales o granulares de inmunoglobulinas, complemento o fibrina en la zona de la membrana basal o en el endotelio vascular en un 70% de los casos16-18. En nuestro estudio solo 3 pacientes (37%) a los que se realizó la IFD presentaron depósitos en la unión dermoepidérmica, 2 de ellos asociaban enfermedad sistémica y presentaban niveles bajos de complemento. Este último hallazgo concuerda con la aseveración por parte de otros estudios de que los casos con IFD positiva están asociados a niveles bajos de complemento en sangre5,6.
Los valores de complemento son uno de los datos de laboratorio clave para el estudio de esta entidad. En nuestra serie destaca el dato de que 7 pacientes (47%) tenían hipocomplementemia, hallazgo superior al encontrado en estudios con una muestra mayor a la nuestra1,5,6,18. El descenso de niveles de complemento normalmente afecta a C1 (C1q), C2, C3 y C4, fracciones que pertenecen a la vía clásica. Son diversos los estudios que aceptan la hipocomplementemia como marcador de enfermedad sistémica1,5,6. En nuestro estudio el 53% de los pacientes presentaba una enfermedad sistémica de carácter autoinmune, lo que aumenta la probabilidad de presentar hipocomplementemia. De hecho, 6 pacientes (88%) de los 7 que presentaban hipocomplementemia tenían enfermedad sistémica asociada, porcentaje muy superior a lo revisado en artículos como el de Mehregan et al.5 y Dincy et al.6. El hecho de que en nuestro caso encontremos un mayor porcentaje de hipocomplementemia y de enfermedad sistémica asociadas se puede atribuir a que la mayoría los pacientes que componen nuestro estudio estaban diagnosticados de una enfermedad sistémica de base previamente, y que estas enfermedades suelen cursar con niveles bajos de complemento. El LES es una de las enfermedades sistémicas a las que se asocia con mayor frecuencia la UV. De hecho, el solapamiento de síntomas entre ambas entidades hace pensar a algunos autores que se encuentren en el mismo espectro de enfermedad19,20.
La UV es una entidad de difícil manejo terapéutico, y a menudo el tratamiento empleado viene marcado por la gravedad de la sintomatología y la enfermedad sistémica a la que esté asociada. Los antihistamínicos son el primer escalón farmacológico para los pacientes con UV y clínica cutánea aislada. También se ha empleado la cinarizina con propiedades antihistamínicas, vasoactivas e inhibidoras de la activación del complemento2. Sin embargo, los antihistamínicos no controlan la inflamación producida por los inmunocomplejos, por lo que es necesaria la utilización de otros fármacos como corticoides orales, indometacina, colchicina, dapsona o hidroxicloroquina, siendo los corticoesteroides los que consiguen una mejoría de la sintomatología en un mayor número de pacientes2,5. En algunos casos, tal y como muestran nuestros resultados, es necesario la utilización de inmunosupresores como la ciclofosfamida y la azatioprina, o fármacos como el rituximab o la inmunoglobulina G intravenosa, fundamentalmente en aquellos casos que asocian una enfermedad sistémica12,21.
ConclusiónEn resumen, las características clínicas encontradas no difieren de estudios previos, presentando los pacientes lesiones con tendencia a durar más de 24h y dejar pigmentación residual. En nuestro caso destaca el elevado porcentaje de casos hipocomplementémicos asociados a una enfermedad sistémica. Por ello, es importante determinar los niveles de complementemia, ya que los si son bajos suelen asociar con mayor frecuencia síntomas extracutáneos y, por ende, traducir una enfermedad sistémica subyacente. Por otro lado, el hecho de tener una enfermedad sistémica de base de carácter autoinmune deberá aumentar el nivel de sospecha de UV en pacientes con lesiones urticariformes sugestivas.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.


