








Mastocytosis is a term used to describe a heterogeneous group of disorders characterized by clonal proliferation of mast cells in various organs. The organ most often affected is the skin. Mastocytosis is a relatively rare disorder that affects both sexes equally. It can occur at any age, although it tends to appear in the first decade of life, or later, between the second and fifth decades. Our understanding of the pathophysiology of mastocytosis has improved greatly in recent years, with the discovery that somatic c-kit mutations and aberrant immunophenotypic features have an important role. The clinical manifestations of mastocytosis are diverse, and skin lesions are the key to diagnosis in most patients.
Las mastocitosis constituyen un grupo heterogéneo de enfermedades caracterizadas por la proliferación clonal de mastocitos en distintos órganos, siendo la localización cutánea la más frecuente. Es «una enfermedad rara o poco frecuente», y afecta a todos los grupos de edad, si bien suele aparecer en la primera década de la vida o entre la segunda y la quinta década de la vida, con una distribución similar por sexos. En los últimos años se han realizado grandes avances en el conocimiento fisiopatogénico del trastorno: las mutaciones somáticas del gen c-kit y la presencia de alteraciones inmunofenotípicas en los mastocitos son elementos importantes en la fisiopatogenia de las mastocitosis. Las manifestaciones clínicas son variadas y las lesiones cutáneas son la clave diagnóstica en la mayoría de los pacientes.
Mastocytosis has an estimated prevalence of 9 cases per 100000 population1 and as such, is considered a rare or uncommon disease according to the European Commission for Public Health, which defines rare diseases as chronically debilitating or life-threatening diseases with a prevalence of under 5 cases per 10000 inhabitants. Two recent epidemiological studies have reported point prevalence rates of 9.2 and 13 cases per 100000 population for indolent systemic mastocytosis (SM) in individuals aged over 15 years.2,3 A similar rate—11.6 cases per 100000 inhabitants—has been described for indolent SM in Hospital General de Albacete in Spain,(unpublished data), and the Instituto de Estudios de Mastocitosis de Castilla-La-Mancha, also in Spain, has estimated an annual incidence of around 0.2 cases per 100000 population for cutaneous mastocytosis.4 The above data, however, are estimates and must be interpreted with caution, as accurate prevalance rates are lacking for solitary mastocytomas and SM without skin involvement associated with anaphylaxis.
Mastocytosis can occur at any age, although onset is more common in the first decade of life. In over 50% of cases the disease appears in the first 2 years of life, with congenital cases being less common.5 Onset is also relatively common between the second and fifth decades of life; males and females are affected similarly.5,6
Family involvement, affecting at least 1 first-degree relative, has been described in 2% to 4% of cases,7,8 most of which have been linked to c-kit germline mutations.9,10
Mastocytosis constitutes a heterogeneous group of disorders that share a common feature, namely the proliferation and accumulation of abnormal mast cells in different tissues, with frequent involvement of the skin, the bone marrow, and the gastrointestinal tract; most patients also develop symptoms secondary to the action of mediators released following mast cell activation.11–14 Mastocytosis is now considered a clonal hematopoietic disease following the demonstration of mutations in the KIT membrane receptor on mast cells in most adult patients15 and in a high proportion of pediatric patients.9
Various forms of mastocytosis exist depending on the age of onset (pediatric or adult forms), the number of affected organs (cutaneous or systemic forms), and clinical behavior (indolent or aggressive forms). Pediatric- and adult-onset forms tend to behave differently. A high proportion of pediatric patients have cutaneous lesions only, with few patients presenting associated symptoms due to the release of mediators from mast cells; furthermore, these lesions tend to disappear around puberty.16–18 Practically 100% of solitary mastocytomas resolve, whereas clinical forms with more extensive lesions can persist in approximately 30% to 50% of cases.8 Patients with adult-onset mastocytosis, by contrast, mostly have systemic involvement (demonstrated by the presence of abnormal mast cells in the bone marrow or at other extracutaneous sites),13,14,19,20 and this form of the disease tends to persist for life. The above classification, however, is questionable, as all forms of mastocytosis originate in the bone marrow, and therefore, at least conceptually, mastocytosis could be considered a systemic disease in all cases.
Pathophysiology of MastocytosisMast cells are hematopoietic cells derived from multipotent myeloid progenitor cells.21 Mast cell precursors migrate from the bone marrow to the blood and then to the tissues, where they terminate their differentiation and acquire the morphologic, immunophenotypic, and functional characteristics of the tissue in which they are located, while maintaining their proliferative ability.11,12
The c-kit proto-oncogene, which is located on chromosome 4q12 in humans,22 encodes KIT (CD117), a surface glycoprotein that acts as a transmembrane receptor with intrinsic tyrosine kinase activity. The KIT protein is expressed in CD34+ hematopoietic bone marrow, peripheral blood, and umbilical cord blood precursors. KIT expression is lost during the maturation of most hematopoietic cells, but not mast cells, where it has a key role in proliferation, survival, and function.23 KIT is also expressed in other cells, including melanocytes and interstitial cells of Cajal in the gastrointestinal tract.4
The KIT receptor structure is organized into 5 domains: a glycosylated extracellular domain, a transmembrane domain, a juxtamembrane membrane, and 2 cytoplasmic domains with tyrosine kinase activity (Fig. 1).23,24 Mast cell precursors mature through activation of the KIT receptor by which the extracellular KIT domain binds to its ligand, stem cell factor, which is essentially synthesized by stromal cells. This interaction between KIT and its ligand has a key role in mast cell development and maturation,25 and it also stimulates adhesion, migration, survival, and the release of mediators by mature mast cells.24
.")
KIT receptor structure.23,24 The location of some activating mutations are shown in red in the juxtamembrane domain and the tyrosine kinase II domain (D816V, which is the most common mutation in mastocytosis).
Mast cells are found in practically all organs and tissues, but they are particularly abundant in the skin, the respiratory system, and the gastrointestinal and genitourinary tracts, especially in the proximity of blood and lymph vessels, and around peripheral nerves. Because mast cells are effector cells of both the innate and acquired immune system, they are located on surfaces near the external medium.26 They are activated by high-affinity immunoglobulin (Ig) E receptors (Fc¿RI) expressed on the surface, but they can also be activated by both immune mechanisms (e.g., IgG receptors [FcγR]27; complement receptors, such as C3aR and C5aR [CD88]28; the high-affinity nerve growth receptor TrKA,28,29 and toll-like or nucleotide-binding oligomerization domain receptors30), and nonimmune mechanisms (drugs and physical triggers31).
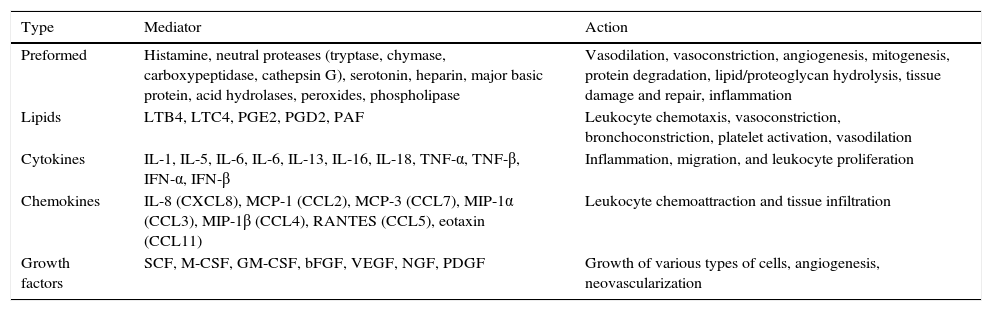
Mast cells are also involved in other functions, such as antigen presentation, angiogenesis, wound healing, tissue remodeling, fibrosis, graft rejection, and tumor surveillance.32 Their activity is induced by the release of multiple mediators, some of which are preformed and stored in granules and others which are synthesized and released in response to the inducing stimulus (Table 1).
Mast Cell Mediators and Physiological Effects.
| Type | Mediator | Action |
|---|---|---|
| Preformed | Histamine, neutral proteases (tryptase, chymase, carboxypeptidase, cathepsin G), serotonin, heparin, major basic protein, acid hydrolases, peroxides, phospholipase | Vasodilation, vasoconstriction, angiogenesis, mitogenesis, protein degradation, lipid/proteoglycan hydrolysis, tissue damage and repair, inflammation |
| Lipids | LTB4, LTC4, PGE2, PGD2, PAF | Leukocyte chemotaxis, vasoconstriction, bronchoconstriction, platelet activation, vasodilation |
| Cytokines | IL-1, IL-5, IL-6, IL-6, IL-13, IL-16, IL-18, TNF-α, TNF-β, IFN-α, IFN-β | Inflammation, migration, and leukocyte proliferation |
| Chemokines | IL-8 (CXCL8), MCP-1 (CCL2), MCP-3 (CCL7), MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5), eotaxin (CCL11) | Leukocyte chemoattraction and tissue infiltration |
| Growth factors | SCF, M-CSF, GM-CSF, bFGF, VEGF, NGF, PDGF | Growth of various types of cells, angiogenesis, neovascularization |
Abbreviations: bFGF, basic fibroblastic growth factor; CCL, chemokine (C-C motif) ligand; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; LTB4, leukotriene B4; LTC4: leukotriene C4; MCP, monocyte chemoattractant protein; M-CSF, macrophage colony-stimulating factor; MIP, macrophage inflammatory protein; NGF, nerve growth factor; PAF, platelet-activating factor; PDGF, platelet-derived growth factor; PG, prostaglandin; RANTES, cytokine expressed and secreted by normal T lymphocytes depending on level of activation; SCF, stem-cell factor; TNF, tumor necrosis factor; VEGF: vascular endothelial growth factor.
Recent years have seen major advances in our understanding of the pathophysiological mechanisms underlying mast cells, and this new information has prompted the development of new diagnostic techniques, treatments, and classification systems. Furthermore, national and European legislative changes have led to the recognition of mastocytosis as a rare or uncommon disease and the publication of monographs. Specialized centers forming part of a broader network have also been created, such as the Spanish Mastocytosis Network (REMA), which is part of the European Consensus Network on Mastocytosis (ECNM).12 These centers draw up consensus documents on diagnosis and treatment to guarantee the right to health for patients with mastocytosis.20
Somatic c-kit mutations and immunophenotypic alterations in mast cells also have a key role in the pathophysiology of mastocytosis.
C-Kit MutationsMultiple mutations have been described as capable of activating the c-kit oncogene receptor independently of its ligand.33,34 The presence of these mutations, known as activating mutations, has been linked to the pathophysiology of gastrointestinal stromal tumors, seminoma, melanoma, and of course, lymphomas, myeloproliferative disorders, and mastocytosis.9,33–35Most mutations implicated in mastocytosis are located in 2 regions of c-kit: exon 11, which encodes the juxtamembrane domain and, more importantly, exon 17, which encodes tyrosine kinase domain II.9 These mutations lead to clonal proliferation via ligand-independent constitutional activation. The most common mutations in this region are missense point mutations in exon 17 (codons 816 and 815); the most common mutation is the substitution of valine for aspartic acid in the catalytic domain of KIT: Asp 816 Val or D816V.11 These mutations have been detected in over 90% of cases of adult mastocytosis.15,33,34 The results for pediatric mastocytosis are more variable (with values ranging from 0%-83%), although it should be noted that only small series focusing on the identification of mutations at codon 816 have been analyzed.33,36–39 Sotlar et al.37 were the first researchers to systematically study the codon 816 mutation in children with pediatric mastocytosis, and they found it to be present in approximately 40% of patients. More recently, Bodemer et al.9 analyzed the c-kit sequence from cutaneous biopsy samples from 50 children aged 0 to 16 years with pediatric mastocytosis. They detected c-kit activating mutations in 86% of patients and found them to be at codon 816 in 42% of cases and in regions outside exon 17 (extracellular and juxtamembrane domains) in 44% of cases. The authors were unable to establish a correlation between type of mutation and phenotype, but they did find an absence of mutations at codon 816 for patients with an onset of disease between the ages of 3 and 16 years. The above studies support the clonal nature of pediatric mastocytosis, despite its tendency to spontaneously regress in many cases.9 Although this hypothesis remains to be confirmed, it will probably be possible one day to identify correlations between different mutations and phenotypes, which would logically have therapeutic repercussions.
In brief, the presence of activating c-kit mutations would appear to be necessary for the development of mastocytosis, while phenotypic diversity could be related to the combination of these mutations with other acquired mutations or inherited genetic polymorphisms.39
The Immunophenotype of Mast Cells in MastocytosisFlow cytometry is capable of identifying and quantifying very low numbers of cells (which is the case of mast cells) and its use in mastocytosis has permitted the detection of a specific immunophenotype for abnormal mast cells in bone marrow and other tissues, namely, the expression of the interleukin α chain receptor CD25.40 The presence of CD25 is an almost pathognomic marker of SM (with the exception of well-differentiated SM),41 and has been associated with cell activation and proliferation.42
CD25 is not found in the mast cells of healthy individuals or, with the exception of FIP1L1/PDGFR alpha- or beta-positive hypereosinophilic syndromes with clonal mast cells, other disorders (hematologic or otherwise).43 More recent studies have evaluated the value of CD30 as a marker for aggressive SM (in which it is expressed in most cases)44 and well-differentiated SM.45
Clinical PresentationThe clinical manifestations of mastocytosis are related to the massive or chronic release of mast cell mediators (which occurs in most pediatric and nonaggressive adult forms),46 tissue infiltration, or the presence of an associated hematologic disorder. The symptoms caused by the release of these mediators include pruritus, reddening, accompanied or not by palpitations and/or headache, blisters arising in skin lesions in certain pediatric forms (Fig. 2), above all in the first years of life, abdominal pain, diarrhea, hypotension, anaphylaxis, and neuropsychiatric symptoms (e.g., irritability, attention deficit).46 In pediatric forms, symptoms tend to be less intense in the 18 months following the appearance of skin lesions.4

Anaphylaxis has been reported in 6% to 9% of cases of pediatric mastocytosis and in 20% to 49% of adult cases.47–49 These ranges are higher than those described for the general population,50,51 but similar to rates reported for IgE-mediated allergic reactions.48,49
Anaphylaxis is common in SM in adults without skin lesions, with a predominance of cardiovascular involvement following hymenoptera venom–induced anaphylaxis in male patients.31,52,53
Sudden onset of symptoms can be triggered by numerous factors, and in particular, physical factors, such as rubbing of lesions, heat, stress, drugs, and wasp stings (Table 2).31
Triggers for the Release of Mast Cell Mediators in Mastocytosis.
| Physical Agents |
| Heat, temperature changes |
| Rubbing against mastocytomas |
| Endoscopy Manipulation of gastrointestinal tract (e.g., during surgery) |
| Emotional factors |
| Stress, anxiety |
| Drugs |
| Nonsteroidal anti-inflammatory drugs |
| Opioids |
| Anesthetics |
| Intravenous iodinated radiocontrast agents Interferon α2b |
| Stings |
| Wasps |
The action of mast cell mediators (histamine, heparin, tryptase, and above all cytokines, such as tumor necrosis factor α, interleukin [IL] 1, and IL-6)54 can lead to bone disorders, such as osteoporosis—detected in approximately 18% of cases of indolent SM55–and diffuse osteosclerosis—detected in 60% of aggressive SM (REMA, unpublished data). A positive correlation has been detected between elevated levels of histamine metabolites in the urine and the risk of osteoporosis.56 The action of mast cell mediators can also give rise to constitutional symptoms, which are seen almost exclusively in aggressive forms of the disease.57
Tissue infiltration, above all in aggressive forms of SM, can give rise to secondary signs and symptoms, such as hepatomegaly and splenomegaly, enlarged lymph nodes, abdominal pain, and altered portal circulation and ascites.13
Although these manifestations are variable, the disease follows an indolent clinical course in most cases. The skin is the most frequently involved organ and is affected in practically 100% of pediatric cases and in around 85% of adult cases. In other words, the absence of skin lesions does not necessarily rule out a diagnosis of mastocytosis.13
The following signs and symptoms can bring a patient with mastocytosis to seek medical attention14:
- 1)
Skin lesions, whether noticed by patients or their families (e.g., in the case of children), or detected during a physical examination for other reasons.
- 2)
Symptoms due to the action of mast cell mediators, such as pruritus, abdominal pain, diarrhea, and anaphylaxis with vascular collapse in the absence of urticaria/angioedema; in some cases the trigger can be a wasp sting.58,59 These symptoms can occur with or without an identifiable trigger and may or may not be IgE-mediated.
- 3)
Asthenia and weight loss, accompanied by hepatomegaly or splenomegaly and blood alterations (anemia, leukocytosis, and thrombocytosis).
- 4)
Pathologic fractures due to advanced osteoporosis in patients without other risk factors for this disease (in particular young or middle-aged men).
- 5)
Nonspecific gastrointestinal symptoms suggestive of colitis or enlarged spleen.
- 6)
Detection of a c-kit mutation in patients undergoing study for a myelodysplastic or myeloproliferative syndrome.14
Skin lesions are the key to diagnosis in many patients, and an expert dermatologist will reach a correct clinical diagnosis of cutaneous mastocytosis in over 90% of cases.4 Noninvasive techniques, such as dermoscopy, can also be of diagnostic value, although findings are nonspecific. Four patterns have been described to date: a uniform brown color, a uniform yellow color, a reticular vascular pattern, and a reticular pigmentation pattern.60 The presence of skin lesions indicates a possible diagnosis of cutaneous mastocytosis, but this must then be confirmed by lesional biopsy20,61,62 with panoptic or metachromatic stains and/or immunohistochemical stains with antibodies against tryptase and/or KIT (Fig. 3). A clinical diagnosis, however, is often sufficient in the case of mastocytomas. Histologic findings include a mastocytic dermal infiltrate with 4 possible patterns: a perivascular pattern in the papillary dermis and upper reticular dermis, a sheet pattern in the upper dermis, an interstitial pattern, and a nodular pattern.63 Cutaneous infiltration by mast cells has no predictive value for systemic involvement, and is only partly correlated with clinical morphology.63
. B, Metachromatic granules in the cytoplasm (toluidine blue, original magnification ×200).")
A definitive diagnosis of cutaneous mastocytosis requires the diagnostic triad of typical skin lesions, histological confirmation of focal mast cell infiltrates in the dermis, and the absence of criteria indicating systemic involvement.13
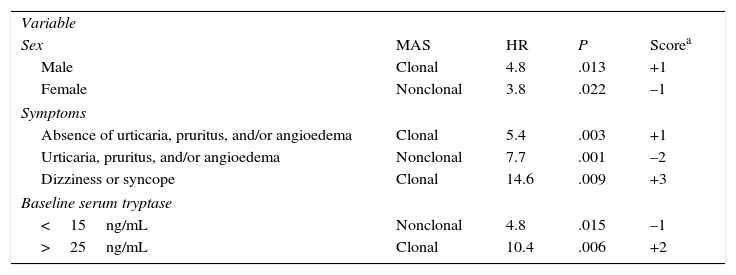
The REMA has designed a special scoring system for patients without typical skin lesions, but in whom mastocytosis is suspected following an episode of anaphylaxis. The system, which has been accepted by the ECNM,64 is used to predict mast cell clonality (KIT mutation [D816V] and/or CD25 expression58) based on clinical and laboratory findings (Table 3).
Spanish Mastocytosis Network Scoring System for Predicting Bone Marrow Mast Cell Clonality and Systemic Mastocytosis in Patients With Symptoms Caused by Mast Cell Activation.
| Variable | ||||
| Sex | MAS | HR | P | Scorea |
| Male | Clonal | 4.8 | .013 | +1 |
| Female | Nonclonal | 3.8 | .022 | –1 |
| Symptoms | ||||
| Absence of urticaria, pruritus, and/or angioedema | Clonal | 5.4 | .003 | +1 |
| Urticaria, pruritus, and/or angioedema | Nonclonal | 7.7 | .001 | –2 |
| Dizziness or syncope | Clonal | 14.6 | .009 | +3 |
| Baseline serum tryptase | ||||
| <15ng/mL | Nonclonal | 4.8 | .015 | –1 |
| >25ng/mL | Clonal | 10.4 | .006 | +2 |
Abbreviations: HR, hazard ratio; c-MAS, clonal mast cell activation syndrome.
The diagnostic criteria for SM as defined by the World Health Organization11,13,14,20 have been evaluated by the REMA in prospective studies. These criteria, designed to raise suspicion of SM, were divided into direct criteria—designed to detect an abnormal anatomic lesion (mast cell aggregates), morphologically abnormal mast cells, expression of surface molecules (CD25), or abnormal molecular markers (c-kit mutations)—and indirect criteria19 (Table 4).
Diagnostic Criteria for Systemic Mastocytosis.
| Direct criteria | Major | Multifocal aggregates of >15 mast cells in bone marrow sections and/or bone marrow smears. These criteria also apply to other tissues. |
| Minor | Atypical morphology in >25% of mast cells in bone marrow smears. | |
| Expression of CD25, with or without CD2, on the surface of bone marrow mast cells. | ||
| Detection of c-kit mutation in exon 17 or elsewhere.a | ||
| Indirect criteria | Elevated baseline serum tryptaseb | |
| Presence of cutaneous mastocytosisc |
A bone marrow study to aid the diagnosis and prognosis of mastocytosis must include cytology; routine histology with classical staining procedures (hematoxylin-eosin), metachromatic stains (e.g., Giemsa or toluidine blue), or more sensitive immunohistochemical staining with antibodies against tryptase (Fig. 4) and the KIT receptor65; flow cytometric analysis of the mast cell immunophenotype11,13,14,20,46,66; and investigation of c-kit mutations in purified bone marrow mast cells and other hematopoietic cell lines.19
.")
As mast cells are bound to the stroma, for a cytological diagnosis to be possible, bone marrow smears must contain a sufficient number of bone marrow particles to permit adequate examination of morphologic features. Mast cells in mastocytosis are elongated and, compared with normal mast cells, they have a hypogranular cytoplasm, an abnormal granular distribution and granular fusion, an ovoid nucleus, and possibly even binucleated cells in more aggressive forms.13
Typical bone marrow lesions are dense (>15 mast cells), multifocal, paratrabecular, or perivascular infiltrates,65 and fibrosis is common in aggressive forms.
The proportion of mast cells in bone marrow is low in both patients with SM (average of 0.27%) and the general population (average of 0.021%).65 The use of flow cytometry for mast cell and immunophenotype detection has undoubtedly advanced knowledge, as this technique provides an enhanced means of detecting, quantifying, and qualifying the pathologic characteristics of mast cells when present in very low numbers.66–68 Purification (to above 97%) of mast cells and other hematopoietic cell lines, such as neutrophils, monocytes, and lymphocytes, using fluorescence-activated cell sorting, can help to establish c-kit pattern mutation patterns (involvement of mast cells only or of other cell lines), which are directly associated with prognosis.15,44,55
The REMA does not recommend routine bone marrow studies in pediatric mastocytosis, except in rare cases when patients develop very serious symptoms following the release of mast cell mediators, with high tryptase levels and associated hepatosplenomegaly and/or cytopenia.
Serum TryptaseTryptases are proteases found in mastocytic granules and, to a lesser extent, blood basophils. Several isoforms have been described in humans, and they are all encoded by genes located in chromosome 16 (e.g., α-tryptase and β-tryptase).
While α-tryptase is released into plasma as a matter of course, β-tryptase is released only following mast cell activation, which occurs in situations associated with massive degranulation of these cells.69
Measurement of total tryptase in plasma or serum has led to one of the greatest breakthroughs in the diagnosis and follow-up of mastocytosis. Tryptase is measured using a commercial immunoassay (ImmunoCAP Tryptase, Thermo Fisher Scientific Inc.) that quantifies total levels in biological fluids; it does not distinguish between mature forms or precursors, or between α and β isoforms.
A baseline serum tryptase level of over 20ng/mL is one of the WHO's minor diagnostic criteria for SM,13 but it should be noted that based on the experience of the REMA, these levels are lower than 20ng/mL in 25% of cases of indolent SM.19
In adults, serum tryptase levels have been associated with total mast cell burden,70 as well as with the extent of bone marrow mast cell infiltration in SM.71 Furthermore, a progressive increase in tryptase levels in serial measurements has been associated with disease progression and worse prognosis.55 This relationship is not so clear in pediatric mastocytosis, however, although elevated tryptase levels have been found in children with extensive cutaneous involvement; these children were also found to have a greater risk of potentially serious symptoms due to the release of mast cell mediators.72
Elevated serum tryptase may also be observed in conditions other than mastocytosis, such as anaphylaxis, myeloid blood disorders, such as leukemia, myelodysplastic syndromes, chronic myeloid leukemia, hypereosinophilic syndromes (characterized by the presence of the FIP1L1/PDGFRA fusion gene and abnormal CD25+ mast cells), and nonhematologic disorders, such as chronic urticaria and advanced kidney failure.55
Additional Tests and Diagnostic AlgorithmsThe following tests are also recommended for the diagnosis and follow-up of mastocytosis: a complete blood count and biochemical analysis, coagulation tests, and histamine metabolites (methylimidazole acetic acid) in urine.73
Additional tests in adults include a bone density test and abdominal ultrasound, although some authors also recommend routine ultrasound in pediatric forms of mastocytosis other than mastocytomas.46 Patients may also be required to undergo a bone scan, computed tomography, or magnetic resonance imaging to check for the presence of enlarged organs, swollen lymph nodes, and/or diffuse or patchy bone sclerosis.
Figs. 5 and 6 show, respectively, a diagnostic algorithm for patients with suspected systemic mastocytosis according to whether or not they have skin lesions.74

Diagnostic algorithm for skin lesions suggestive of mastocytosis.
Source: Alvarez-Twose et al.58
 scoring system (see Table 3). Source: Valent et al.64")
Although mastocytosis is a rare or uncommon clonal disease with varying manifestations, it follows an indolent clinical course in most cases. The skin is the most frequently affected organ. Systemic involvement should be routinely investigated by bone marrow studies in adults and in children when there is a high index of clinical suspicion.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
We thank Dr Luis Escribano for his leadership, motivation, and commitment to the study of mastocytosis.
Please cite this article as: Azaña JM, Torrelo A, Matito A. Actualización en mastocitosis. Parte 1: fisiopatología, clínica y diagnóstico. Actas Dermosifiliogr. 2016;107:5–14.


