









Mastocytosis is a term used to describe a heterogeneous group of disorders characterized by clonal proliferation of mast cells in different organs. The organ most often affected is the skin. The World Health Organization classifies cutaneous mastocytosis into mastocytoma, maculopapular cutaneous mastocytosis, and diffuse mastocytosis. The systemic variants in this classification are as follows: indolent systemic mastocytosis (SM), aggressive SM, SM with an associated clonal hematological non-mast cell lineage disease, mast cell leukemia, mast cell sarcoma, and extracutaneous mastocytoma. The two latest systemic variants are rare. Although the course of disease is unpredictable in children, lesions generally resolve by early adulthood. In adults, however, the disease tends to persist. The goal of treatment should be to control clinical manifestations caused by the release of mast cell mediators and, in more aggressive forms of the disease, to reduce mast cell burden.
Las mastocitosis constituyen un grupo heterogéneo de enfermedades caracterizadas por la proliferación clonal de mastocitos en distintos órganos, siendo la localización cutánea la más frecuente. La Organización Mundial de la Salud (OMS) clasifica las mastocitosis cutáneas en mastocitomas, mastocitosis máculo-papulosas y mastocitosis cutánea difusa, mientras que las formas sistémicas incluyen las mastocitosis indolentes, las agresivas, las asociadas a otra hematopatía monoclonal y la leucemia mastocitaria; el sarcoma mastocitario y el mastocitoma extracutáneo son variantes muy poco frecuentes. Aunque la evolución de la enfermedad en los niños es impredecible, con frecuencia las lesiones desaparecen durante la infancia; en los adultos la enfermedad tiende a persistir. El tratamiento se dirige a controlar las manifestaciones clínicas debidas a la acción de los mediadores mastocitarios, mientras que las formas agresivas requerirán de tratamientos dirigidos a reducir la masa mastocitaria.
Mastocytosis is classified into different categories of disease based on World Health Organization (WHO) criteria and consensus proposals (Table 1).1–4 To discuss these categories, we have divided them into cutaneous and systemic forms.
Classification of Mastocytosis.
| Cutaneous mastocytosis | Urticaria pigmentosa | |
| Diffuse cutaneous mastocytosis | ||
| Mastocytoma of skin | ||
| Systemic mastocytosis | Indolent systemic mastocytosis | |
| Smouldering (latent) systemic mastocytosisa | ||
| Mastocytosis with an associated monoclonal hematologic disease | Myelodysplastic syndrome Myeloproliferative syndrome Acute myeloid leukemia Non-Hodgkin lymphoma | |
| Aggressive systemic mastocytosis | ||
| Mast cell leukemia | ||
| Mast cell sarcoma | ||
| Extracutaneous mastocytoma | ||
From a dermatologic perspective, there is still no ideal classification system that correlates lesion type with prognosis, i.e., the likelihood of the disease regressing or progressing to systemic involvement. Nonetheless, 5 morphologically distinct types of cutaneous lesions have been described in mastocytosis.5,6
- 1.
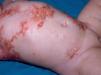
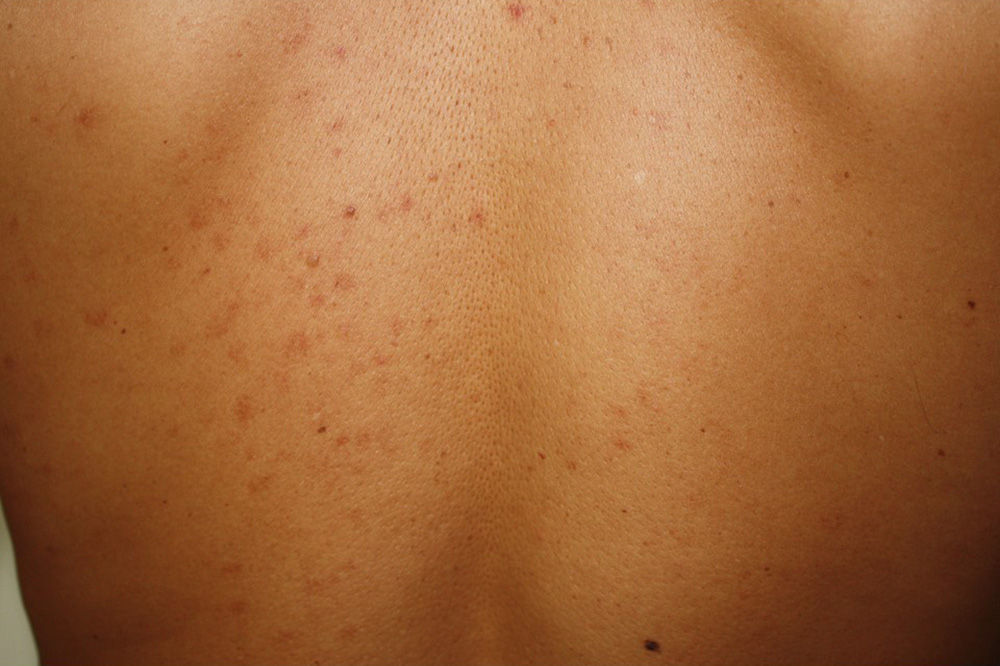
Maculopapular cutaneous mastocytosis. Maculopapular lesions are the most common clinical presentation of mastocytosis and have been traditionally been included in the category of urticaria pigmentosa. They are characterized by red, tan, or brown macules and papules that vary in size and number and are located preferentially on the trunk2,7 (Fig. 1). The lesions tend to persist in adults, but in two-thirds of children, they regress either partially or completely.
- 2.
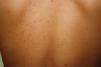
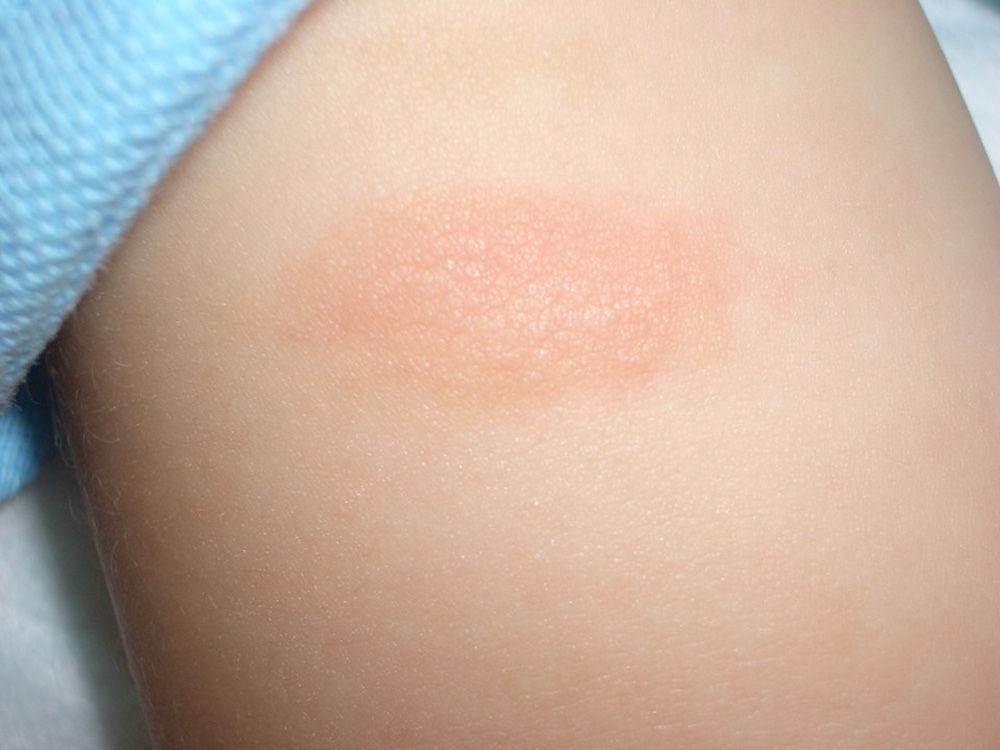
Plaque-type lesions are typical in pediatric mastocytosis and generally have a favorable prognosis (Fig. 2).
- 3.
Mastocytoma or nodular mastocytosis (single lesion or various lesions) (Fig. 3). Solitary cutaneous mastocytoma is characteristic of pediatric mastocytosis, although isolated cases have been described in adults.9,10 In one series of 33 children, 60% of mastocytomas were found to be congenital and over half were located on the extremities, but sparing the palms and soles.11 The clinical manifestations tend to be cutaneous and confined to the lesion or lesions involved; they include mild pruritus, blisters (reported only in infants and young children), and urticaria caused by rubbing or friction. A positive Darier sign12 is diagnostic of mastocytosis, but friction can also induce acute systemic lesions due to the release of mast cell mediators, particularly in larger lesions.11 Mastocytomas are benign and tend to disappear during childhood.2,11
- 4.
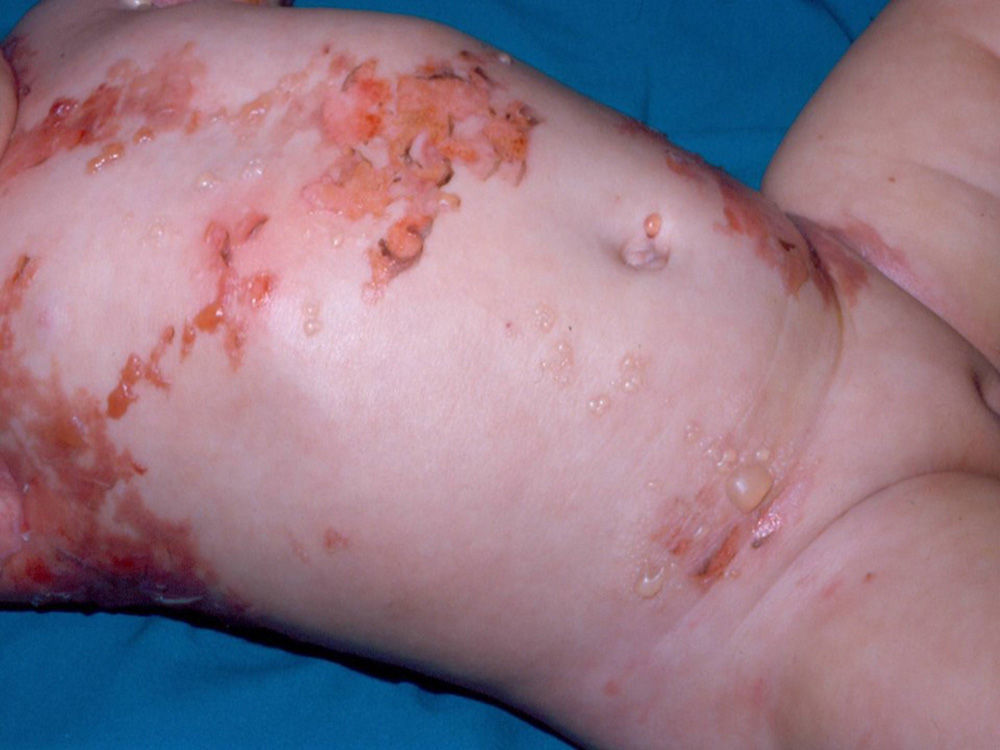
Diffuse cutaneous mastocytosis. This is an uncommon form of cutaneous mastocytosis. It appears in neonates or in the first months of life and is characterized by intense symptoms due to increased cutaneous infiltration by mast cells. Blisters, which may be hemorrhagic, are generally the first manifestation, and hence the differential diagnosis at this age is very broad (Fig. 4). In the 18 months following onset, symptoms induced by the release of mast cell mediators tend to be severe and even life-threatening–but they generally subside over time.8,13
The term diffuse cutaneous mastocytosis has been used to refer to mast cell lesions with extensive cutaneous involvement, and as such, it encompasses maculopapular lesions and/or very extensive nodular lesions, as well as generalized erythrodermic forms.14 This term, however, should be reserved for erythrodermic mastocytosis.14
- 5.
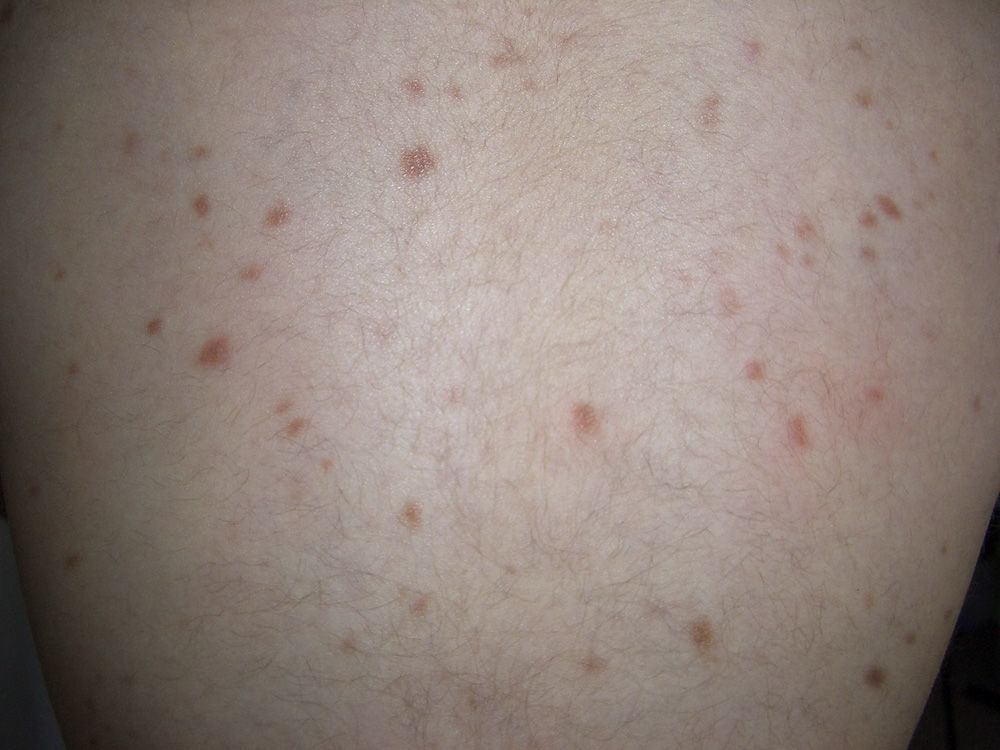
Telangiectatic cutaneous mastocytosis. This form occurs primarily in adults, and is only occasionally observed in children. It is characterized by red and/or brown telangiectatic macules located predominantly on the trunk (Fig. 5). In this classification, telangiectatic cutaneous mastocytosis is included as a separate entity, rather than as a variant of maculopapular cutaneous mastocytosis. In previous classifications, it was termed telangiectasia macularis eruptiva perstans.4,5
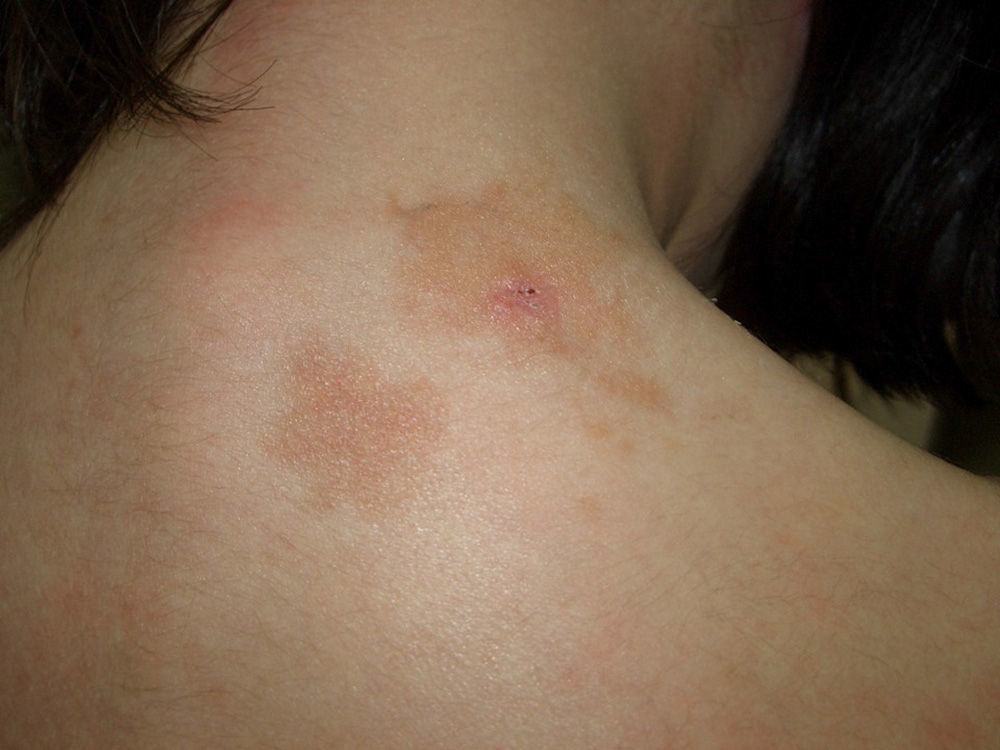
As is the case with other skin disorders, clinical presentations with skin lesions confined to a particular segment or area of the body have been described as nevoid, linear, or unilateral.15–17 Based on the work of Happle,18 these forms would constitute a mosaic manifestation (type 1 mosaicism) of the disease, in which the mutational event, albeit unconfirmed, would be limited to the part of the body affected. These forms are rare.
The above classification of cutaneous mastocytosis has considerable limitations in terms of clinical presentation (pediatric lesions are more heterogeneous than adult lesions) and prognosis (tendency to spontaneously regress or progress to systemic disease), and is currently under revision.
Systemic MastocytosisIndolent systemic mastocytosis (ISM) is the most common presentation of SM in adults, and can present with skin lesions (ISM[+]) or without (ISM[-]). ISM(+) accounts for 79% of SM cases being monitored by the Spanish Mastocytosis Network (REMA)19 while ISM(-)20 can occur in association with anaphylaxis with predominantly cardiovascular symptoms.
The WHO has defined a set of criteria–referred to as B findings and C findings–to establish the aggressive nature of mastocytosis (Table 2).1,2,4 While IMS does not have either B or C findings, the WHO classification includes a category known as smouldering mastocytosis, which is a latent systemic form characterized by 2 or more B findings and no C findings.
World Health Organization Criteria to Establish the Aggressive Nature of Mastocytosis.
| B Findings |
| High mast cell burden with bone marrow infiltration >30% and tryptase levels >200ng/mL |
| Hypercellular bone marrow or signs of myelodysplasia or stable changes in peripheral blood counts |
| Hepatomegaly and/or splenomegaly without functional alterations; lymph nodes > 2cm on ultrasound or computed tomography scan |
| C Findings |
| Altered organ function |
| ≥1 cytopenias Leukocytes <1000/μL, hemoglobin <10g/dL, platelets <100000/μL |
| Hepatosplenomegaly with ascites, altered liver function, or portal hypertension Palpable spleen with hypersplenism |
| Intestinal malabsorption with hypoalbuminemia and weight loss |
| Bone lesion with osteolysis and/or severe osteoporosis with pathologic fracturesa |
Patients with ISM(-) have a lower bone marrow mast cell burden than those with ISM(+), and the number of patients with mast cell aggregates is also lower. In most cases of ISM(-), the mast cells show an activated immunophenotype and a different mutation pattern, as c-kit mutations other than the typical D816V mutation mentioned in part 1 of this review are found in exon 17 in 6% of cases. Furthermore, the percentage of patients harboring c-kit mutations in several cell lines (risk factor for disease progression) is much lower in ISM(-) than in ISM(+) (6% vs 23%).20
Well-differentiated SM21 is another uncommon form of mastocytosis that accounts for 6% of all cases of adult SM (REMA, unpublished data). Onset occurs in childhood in the vast majority of cases, and patients develop nodular cutaneous lesions of varying size that predominantly affect the trunk, neck, and upper portion of the limbs, in addition to symptoms induced by the release of mast cell mediators. Bone marrow mast cells are large, round and have a central nucleus and abundant, regularly distributed granules. Cytoplasmic vacuoles are common. These cells do not express the CD25 antigen and their immunophenotype is mature, like that of normal mast cells; they do, however, tend to express CD30.3,22C-kit mutations are found in exon 17 in a very low proportion of cases (29%),23 although there have been isolated reports of mutations in other regions of the c-kit gene (e.g., the transmembrane mutation Phe522Cys).21
Aggressive SM accounts for 6% of all cases of SM monitored by the REMA, and 60% of patients have skin lesions (REMA, unpublished data). Manifestations include considerable hepatosplenomegaly, abdominal lymph nodes larger than 2cm, frequent ascites and pleural effusion, severe malabsorption with hypoproteinemia and hypoalbuminemia, cytopenia due to hypersplenism, bone marrow infiltration, and fibrosis, or a combination of the aforementioned. Tryptase levels are in excess of 200ng/mL. Histologic examination of bone marrow smears shows marked mast cell infiltration, CD25+ mast cells with an immature immunophenotype,24 and c-kit mutations affecting other hematopoietic cell lines, with myeloid and lymphoid cells involved in 50% of cases.25
Mast cell leukemia is a rare form of SM in which mast cells account for 20% or more of all nucleated cells and for at least 10% of all blood cells in peripheral blood (leukemic mast cell leukemia). When mast cells represent less than 10% of these blood cells, the condition is called aleukemic mast cell leukemia.3
Mast cell sarcoma (a solid tumor with a poor prognosis) and extracutaneous mastocytoma are very rare clinical forms of mastocytosis.3
PrognosisPrognosis is generally favorable in pediatric mastocytosis, with most cases resolving around puberty. However, the number of cases that persist into adulthood might be underestimated, as little information is available on such cases.8,26–28
Solitary lesions typically disappear before adulthood.8,29–31 Multiple nodular lesions can also resolve spontaneously, but when they persist into adulthood, patients tend to experience extensive skin involvement and symptoms caused by the release of mast cell mediators. This presentation is probably well-differentiated SM,14 and as occurs in diffuse cutaneous (erythrodermic) mastocytosis, in which symptoms improve with time, the skin tends to acquire a hyperpigmented, thickened appearance (sometimes referred to as elephant skin), and bone marrow studies often reveal well-differentiated matsocytosis.13
Systemic mastocytosis with childhood onset persists into adulthood,32 and there have also been isolated reports of pediatric mastocytosis with a fatal outcome following progression to an aggressive systemic form.8
As no predictive factors have yet been described in pediatric mastocytosis, long-term monitoring is required, as are prospective studies to evaluate the prognostic significance of different clinical and laboratory findings.
Adult-onset mastocytosis tends to persist. In indolent SM, which is the most frequent presentation, the likelihood of progression to more aggressive disease is directly related to the pattern of c-kit mutations. In forms with mutations affecting mast cells only, the probability of progression at 30 years is 0%, while in forms with mutations affecting several cell lines, it is 1.7% (SD, 1.2%) at 10 years and 8.4% (SD, 5%) at 20 and 25 years.24,33
TreatmentAs no cures are currently available for mastocytosis and the prognosis is generally favorable, the goal of treatment is symptomatic relief in most cases. Treatment recommendations include general measures aimed at preventing factors that can trigger massive mast cell degranulation and more specific measures to control symptoms caused by the acute or chronic release of mast cell mediators; cytoreductive therapy is also recommended in aggressive mastocytosis to reduce the mast cell burden.34,35
General MeasuresAs in other rare or uncommon diseases, the management of mastocytosis should involve multidisciplinary teams and contact with specialized reference centers.
All patients and/or carers should be provided with written information on the disease, including general information on factors that can trigger symptoms due to the release of mast cell mediators, and personalized information adapted to the patient's history of tolerance of these triggers. The Instituto de Estudios de Mastocitosis de Castilla-La Mancha, for instance, has produced a patient information sheet on mastocytosis that is available (Specific protocols are available in several idioms) on its website (www.mastocitosis.org) and on the website of the Spanish Mastocytosis Association (www.mastocitosis.com).
Treatment of Pediatric MastocytosisPhysical measures can have a very important impact on reducing mast cell activation and the need for pharmacologic treatment.36 These measures include avoidance of sudden changes in temperature, exposure to heat, and rubbing of lesions.
Treatment should be individualized according to the type, severity, and frequency of manifestations (Table 3) and should also be implemented in a stepwise fashion.
Pharmacologic Treatment of Signs and Symptoms Secondary to the Release of Mast Cell Mediators in Pediatric Mastocytosis.
| General measures | Avoidance of triggers Avoidance of sudden changes in temperature Avoidance of rubbing of lesions |
| Topical treatment | Hydration Disodium cromoglycate (0.21%-4% every 6-8h) Topical corticosteroids Pimecrolimus |
| Specific treatment for mastocytoma | Local measures/topical treatment Surgical excision where appropriate |
| Specific treatment for maculopapular mastocytosis | General measures H1 antihistamines (sedating/nonsedating) H2 antihistamines Oral disodium cromoglycate (15-20 mg/kg/d divided into 3 doses, to be taken on an empty stomach) Antileukotrienes (montelukast) Photochemistry (psoralens and UV-A, UV-B) |
| Specific treatment for diffuse cutaneous mastocytosis | General measures Management in a specialized unit/intensive care where appropriate The above treatments at full doses Photochemotherapy |
Topical treatments include disodium cromoglycate at a concentration of 0.21% to 4% in aqueous solutions.36,37 Topical corticosteroids applied to open wounds or using occlusive overnight dressings have also proven effective,36,37 as have intralesional corticosteroid injections.38 Sedating and nonsedating H1 antihistamines administered on a continuous or on-demand basis, in addition to H2 antihistamines are also used. Oral cromoglycate (15-20mg/kg/d) is prescribed when symptoms are not relieved by antihistamines, if the patient develops abdominal pain, diarrhea, irritability, or sleep disorders, or if an unexplained reduction is observed in cholesterol, triglyceride, ferritin, or vitamin B12 levels are observed.36 The mechanism of action of oral cromoglycate is not fully understood as its absorption in minimal. Leukotriene antagonists are also administered to patients with difficult-to-control symptoms.38 Phototherapy with psoralen combined with either UV-A or UV-B is reserved for rare cases of massive cutaneous involvement, where patients experience severe symptoms and repeated formation of blisters and other symptoms caused by the massive release of mast cell mediators that do not respond to the above treatments.14,34,36
Diffuse cutaneous mastocytosis, which is very rare, should be considered a true medical emergency due to the high risk of widespread mast cell release, and patients require admission to specialized units experienced in this condition and the use of full-dose treatments, including phototherapy.36
Treatment with chemotherapeutic agents such as interferon and tyrosine kinase inhibitors, which modulate biologic response, is formally contraindicated in children with mastocytosis, except in life-threatening situations.34,38
Treatment of Adult MastocytosisIn addition to general measures, all patients should be administered treatment aimed at controlling symptoms due to mast cell mediator release in accordance with the type and severity of symptoms they typically experience.35 Different combinations of drugs are indicated depending on the patient's clinical manifestations (Table 4). Patients are generally treated with oral disodium cromoglycate, H1 antihistamines (administered on a scheduled or on-demand basis), and H2 antihistamines. Systemic corticosteroids are used in cases that do not respond to the above treatments and patients should also be given self-injectable adrenaline for anaphylactic emergencies.35,39
Pharmacologic Treatment of Signs and Symptoms Secondary to the Release of Mast Cell Mediators in Adult Mastocytosis.
| Signs and Symptoms | Drug |
|---|---|
| Cutaneous | H1 and H2 antihistamines Topical disodium cromoglycate (0.21%-4% every 6-8h) Oral disodium cromoglycate (200mg every 6h on an empty stomach) Topical corticosteroids Oral corticosteroidsa |
| Gastrointestinal | Oral disodium cromoglycate H2 antihistamines and proton pump inhibitors Oral corticosteroids or cyclooxygenase 2 inhibitorsb |
| Cardiovascular | Oral disodium cromoglycate H1 and H2 antihistamines Antileukotrienes (montelukast) Oral corticosteroids/acetylsalicylic acidb Self-injectable epinephrinec |
| Anaphylaxis | Self-injectable epinephrine Systemic corticosteroids H1 antihistamines Oral disodium cromoglycate H1 and H2 antihistamines Antileukotrienesd Omalizumabe |
| Bone marrow deficit | Calcium and vitamin D Bisphosphonates Interferon alfa 2be |
Patients in whom anxiety and/or stress trigger symptoms due to the release of mast cell mediators should be referred for psychiatric and psychological evaluation and administered anxiolytics and/or antidepressants where appropriate.
Hymenoptera specific immunotherapy is recommended in patients who have experienced an anaphylactic episode triggered by immediate hypersensitivity to wasp venom; calcium supplements, vitamin D, and even bisphosphonates are indicated on detection of bone marrow deficits.35
Anti-immunoglobulin E therapy with omalizumab has been used successfully in mastocytosis and is reserved for patients with severe symptoms secondary to mediator release that are refractory to treatment with conventional antimediator therapy.40 The efficacy of omalizumab in mastocytosis is currently being evaluated in a controlled clinical trial setting.38
In advanced forms of the disease with a high mast cell burden, cytoreductive therapy with hydroxyurea, interferon alfa 2b, or cladribine (2-CdA) is recommended.38
Tyrosine kinase inhibitors may be a promising alternative for the treatment of systemic mastocytosis. However, as things stand, only imatinib has proven useful in the few cases of mastocytosis in which the c-kit mutation is found outside the kinase tyrosine domain II, which harbors the typical D816V mutation, as patients with this mutation do not respond to imatinib. Other tyrosine kinase inhibitors such as masitinib and dasatinib have been used in very few cases, and have produced modest results in terms of the reduction of bone marrow mast cell burden. Finally, midostaurin (PKC412) has been used to treat advanced systemic mastocytosis, with or without the c-kit D816V mutation.38
ConclusionsMastocytosis is a clonal disease with varying clinical manifestations and c-kit receptor mutations present in a high proportion of cases. Pediatric mastocytosis tends to be limited to the skin, is indolent, and often resolves before adulthood. Adult forms tend to persist and are characterized by systemic involvement in over 90% of cases, although the course is generally indolent. The goal of treatment is typically to relieve symptoms caused by the release of mediators from mast cells, ensure avoidance of triggers, control symptoms using scheduled or on-demand antimediator therapy, and reduce mast cell burden in patients with advanced SM.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
We thank Dr Luis Escribano for his leadership, motivation, and commitment to the study of mastocytosis.
Please cite this article as: Azaña JM, Torrelo A, Matito A. Actualización en mastocitosis. Parte 2: categorías, pronóstico y tratamiento. Actas Dermosifiliogr. 2016;107:15–22.


