

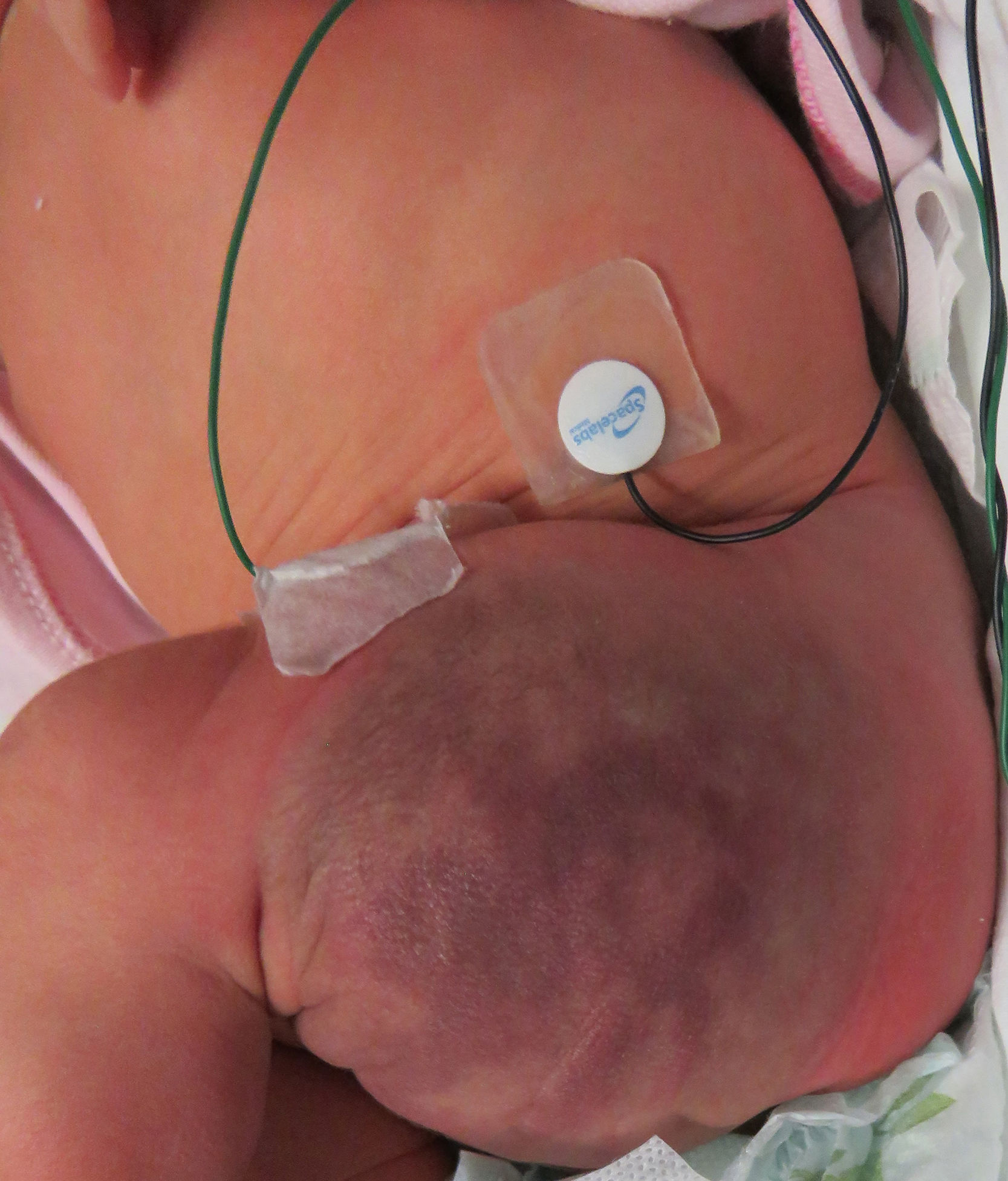
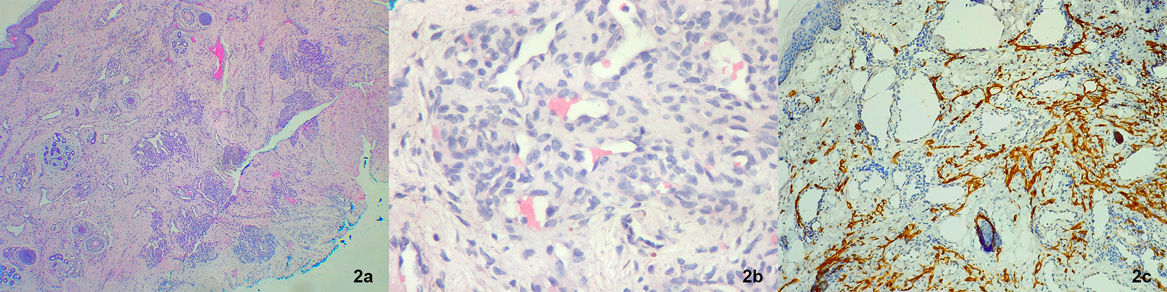
Una recién nacida a término, fruto de embarazo controlado, sin complicaciones, fue evaluada a los 3 días de vida por un tumor vascular congénito localizado en el miembro inferior izquierdo, sin otros antecedentes de relevancia. Al examen físico presentaba una tumoración blanda de 8×6cm, eritemato-violácea, con centro purpúrico, halo pálido periférico y zonas de hipertricosis, localizada en el muslo izquierdo (fig. 1). En la ecografía Doppler de la lesión se observó un aumento del espesor y de la ecogenicidad del tejido subcutáneo, con imágenes vasculares dilatadas que se extendían hasta el plano muscular. En el ecocardiograma Doppler no se observaron signos de sobrecarga cardíaca. El análisis de sangre mostró un perfil hematológico sin alteraciones, y las ecografías abdominal y cerebral no tenían hallazgos patológicos. El estudio histopatológico de la tumoración evidenció una proliferación de células fusiformes que, por sectores, se disponían formando nódulos y delimitando capilares. Algunos de los nódulos estaban rodeados por un vaso de mayor tamaño en forma de semiluna y, en otros sectores, se disponían en islotes sólidos. Se realizaron técnicas de inmunohistoquímica que fueron positivas para D2-40, CD31 y WT1, siendo GLUT1 negativo (fig. 2).

 Imagen en «bolas de cañón» (H&E, ×4); b) Proliferación de células fusiformes que se disponen en islotes sólidos en la profundidad (H&E, ×40); c) Inmunohistoquímica D2-40: positivo.")
¿Cuál es el diagnóstico?
DiagnósticoAngioma en penacho/hemangioendotelioma kaposiforme.
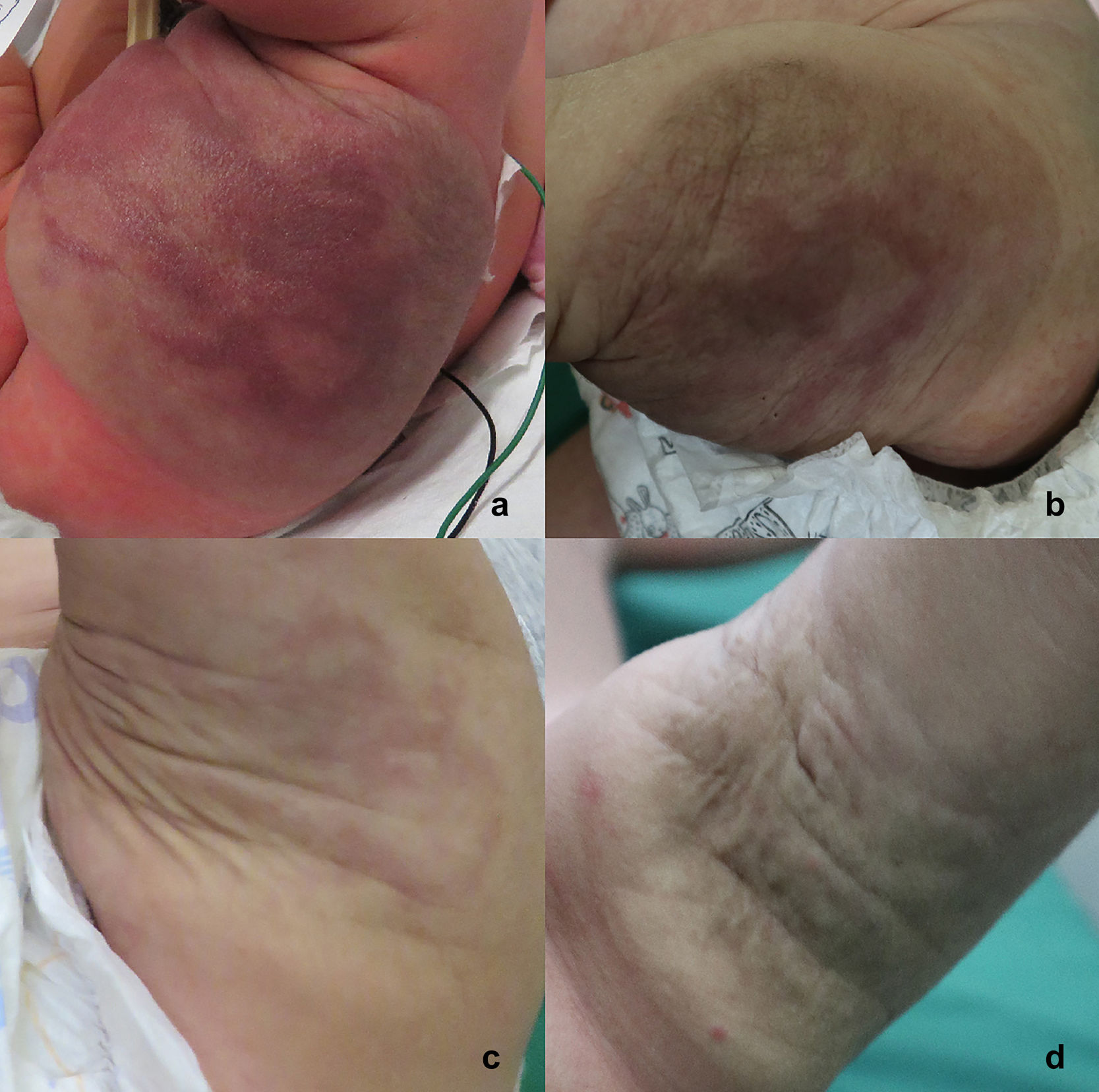
Evolución y comentarioLos hallazgos clínico-patológicos permitieron establecer el diagnóstico de angioma en penacho/hemangioendotelioma kaposiforme. En su evolución, la paciente presentó disminución progresiva del tamaño de la lesión (fig. 3), sin alteraciones hematológicas asociadas tras 11 meses de seguimiento; razón por la cual se decidió realizar un estricto control clínico de forma interdisciplinaria con pediatras, dermatólogos y radiólogos, sin tratamiento farmacológico.
 3 días de vida; b) 18 días de vida; c) 6 meses de edad; d) 11 meses de edad.")
El angioma en penacho (AP) y el hemangioendotelioma kaposiforme (HEK) son tumores vasculares poco frecuentes, clasificados por la International Society for the Study of Vascular Anomalies (ISSVA) como benigno y localmente agresivo, respectivamente. En la actualidad, debido a la similitud en las características histopatológicas y su capacidad para desarrollar el fenómeno de Kasabach Merritt (FKM), algunos autores consideran que estas entidades forman parte de un mismo espectro de enfermedad1.
Presentan una leve predominancia por el sexo masculino2,3. Se los ha asociado a mutaciones genéticas de la familia GNAQ y en algunos casos, a mutaciones en GNA144.
Los AP se presentan como placas o tumores eritematovioláceos en niños y adultos jóvenes, siendo infrecuente su presencia al nacimiento1,5. Pueden asociarse a hiperhidrosis o hipertricosis y suelen localizarse en miembros inferiores. Algunas lesiones involucionan de forma espontánea, especialmente los casos de presentación congénita1,5,6, como el caso de nuestra paciente. Los HEK se presentan como placas o tumores eritematovioláceos, los cuales pueden comprometer la piel de forma superficial o profunda, comportándose como tumores localmente agresivos ya que pueden afectar tejidos en profundidad1,7. Se localizan principalmente en las extremidades y, en menor frecuencia, en tronco, retroperitoneo, cabeza y cuello, mediastino y órganos como timo y bazo2,3,7. Suelen presentarse durante el primer año de vida, siendo aproximadamente la mitad de las lesiones evidentes al nacimiento2,3. La presencia de complicaciones es más frecuente en pacientes con HEK, pudiendo encontrarse FKM, dolor, limitación funcional, linfedema y compresión de estructuras vitales como la vía aérea2,3. El FKM se observa hasta en un 70% de los pacientes con HEK y en un 10% con AP3,7. Se caracteriza por trombocitopenia severa y coagulopatía por consumo, que puede llegar a comprometer la vida del paciente2,3,7. Se postulan como factores de riesgo la temprana edad (menores de 6 meses), localización en tronco, tamaño mayor a 5cm y lesiones profundas3, como presentaba nuestra paciente. El diagnóstico se realiza por las manifestaciones clínicas confirmándose por el estudio histopatológico2. En la histopatología puede observarse un infiltrado de células endoteliales fusiformes que se disponen en nódulos bien definidos y redondeados, que se alinean para formar canales linfáticos y estructuras vasculares en forma de semiluna. Esta imagen se la conoce con el nombre de «patrón en bolas de cañón», característico del AP. En el caso del HEK, estas mismas células endoteliales fusiformes se disponen formando playas sólidas2,6.
La inmunohistoquímica muestra positividad para marcadores endoteliales vasculares (CD31 y CD34), linfáticos (D2-40, LYVE-1, Prox-1 y VEGFR-3) y negatividad para GLUT-12,6. Como diagnósticos diferenciales se plantean al hemangioma infantil, hemangioma congénito, malformaciones venosas y linfangiomatosis kaposiforme, entre otros2. El tratamiento del AP/HEK depende del tamaño de la lesión, profundidad, evolución y presencia o no de complicaciones. Como opciones terapéuticas encontramos los procedimientos quirúrgicos, el tratamiento farmacológico7 y, en ocasiones, la observación clínica. Como tratamiento farmacológico se pueden utilizar corticoides, vincristina y sirolimus (0,8mg/m2/día VO, buscando unos niveles plasmáticos entre 5 y 10μg/dl si no presenta coagulopatía o entre 10 a 15μg/dl si la presenta)2,6–8.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



