



Ataxia-telangiectasia (A-T), or the Louis-Bar syndrome is a rare autosomal recessive disease characterized by cerebellar degeneration, the appearance of cutaneous telangiectasias, immunodeficiency, premature ageing, sensitivity to radiation, and early-onset neoplasms. For all these reasons, it is often categorized as a disorder of genomic instability and DNA repair defects.1–4
This disease is due to a mutation in the ATM gene that leads to dysfunction of the ATM kinase protein, which is responsible for regulating the cell cycle by activating various proteins such as BRCA1 or p53, among others.1–4
Patients often present with neurological signs, including difficulty walking, abnormal eye movements, cognitive impairments, and speech difficulties.1,3,4
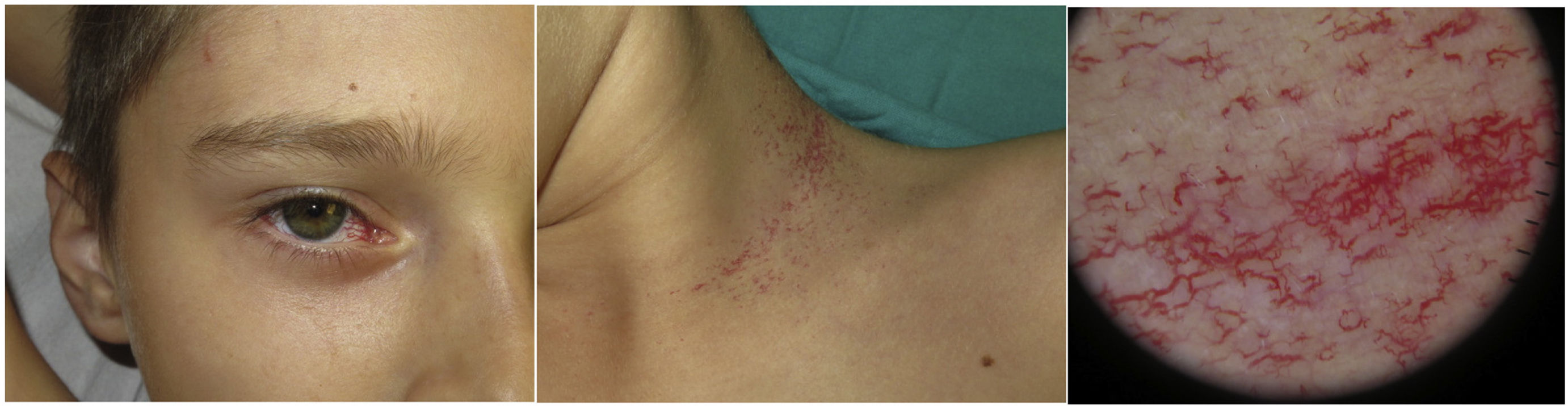
Skin abnormalities often appear between the ages of 3 and 6 years and the following are very typical of this condition (Fig. 1)1,3,4:
- -
Bulbar conjunctival telangiectasias are the most distinctive finding. They can also be seen in exposed areas of the body, especially the facial and cervical regions.
- -
Patients can show café-au-lait spots, hypopigmented macules, melanocytic nevi, chronic cutaneous granulomas of torpid evolution (related to rubella vaccination, which is contraindicated in these patients), papulosquamous facial rash, vitiligo, and premature ageing.
These patients also experience endocrine dysfunction (growth delay, insulin resistance, diabetes, and delayed puberty), humoral and cellular immunodeficiency, and frequent sinopulmonary infections. Early-onset neoplasms, especially of haematological origin, contribute to the high morbidity and mortality rates and grim prognosis reported, with a median survival age of 25 years, since there are no drugs to date that can change the course of the disease.1,3
A-T has been considered a model for studying ageing in humans. Recently, an interesting article published in Ageing Research Reviews by Aguado et al.2 showed nine mechanisms that contribute to this premature ageing: genomic instability, impaired intercellular communication, telomere shortening, cellular senescence, epigenetic changes, oxidative stress and mitochondrial dysfunction, loss of proteostasis, depletion of stem cells, and deregulated nutrient sensing.
In addition to detailing the genetic and molecular mechanisms underlying premature ageing, the article illustrates the main treatments currently in the pipeline, highlighting emerging therapeutic opportunities that can improve the phenotypes associated with premature ageing in A-T.
In conclusion, the importance of dermatological and ocular findings for the diagnosis of A-T in a patient with neurological deterioration is emphasized in this article, as well as the advances that can be made in the understanding of human biology by studying the genetics of rare diseases, and the potential therapeutic advancements that can result from such research.


