





Atopic dermatitis is the most common chronic inflammatory skin disorder, affecting up to 20% of children and 10% of adults in developed countries. The pathophysiology of atopic dermatitis is complex and involves a strong genetic predisposition and T-cell driven inflammation. Although our understanding of the pathology and drivers of this disease has improved in recent years, there are still knowledge gaps in the immune pathways involved. Therefore, advances in new omics technologies in atopic dermatitis will play a key role in understanding the pathogenesis of this burden disease and could develop preventive strategies and personalized treatment strategies. In this review, we discuss the latest developments in genetics, transcriptomics, epigenomics, proteomics, and metagenomics and understand how integrating multiple omics datasets will identify potential biomarkers and uncover nets of associations between several molecular levels.
La dermatitis atópica es el trastorno inflamatorio de la piel crónico más común. Afecta hasta a 20% de los niños y a 10% de los adultos en países desarrollados. La fisiopatología de la dermatitis atópica es compleja e implica una fuerte predisposición genética e inflamación impulsada por células T. Aunque nuestra comprensión de la patología y las causas de esta enfermedad ha mejorado en los últimos años, aún existen lagunas de conocimiento en las vías inmunológicas involucradas. En consecuencia, los avances en nuevas tecnologías ómicas en la dermatitis atópica desempeñarán un papel clave en la comprensión de la patogénesis de esta enfermedad y podrían desarrollar estrategias preventivas y tratamientos personalizados. En esta revisión se discuten los últimos avances en genética, transcriptómica, epigenómica, proteómica y metagenómica, y entendemos cómo la integración de múltiples conjuntos de datos ómicos identificará posibles biomarcadores y descubrirá redes de asociaciones entre varios niveles moleculares.
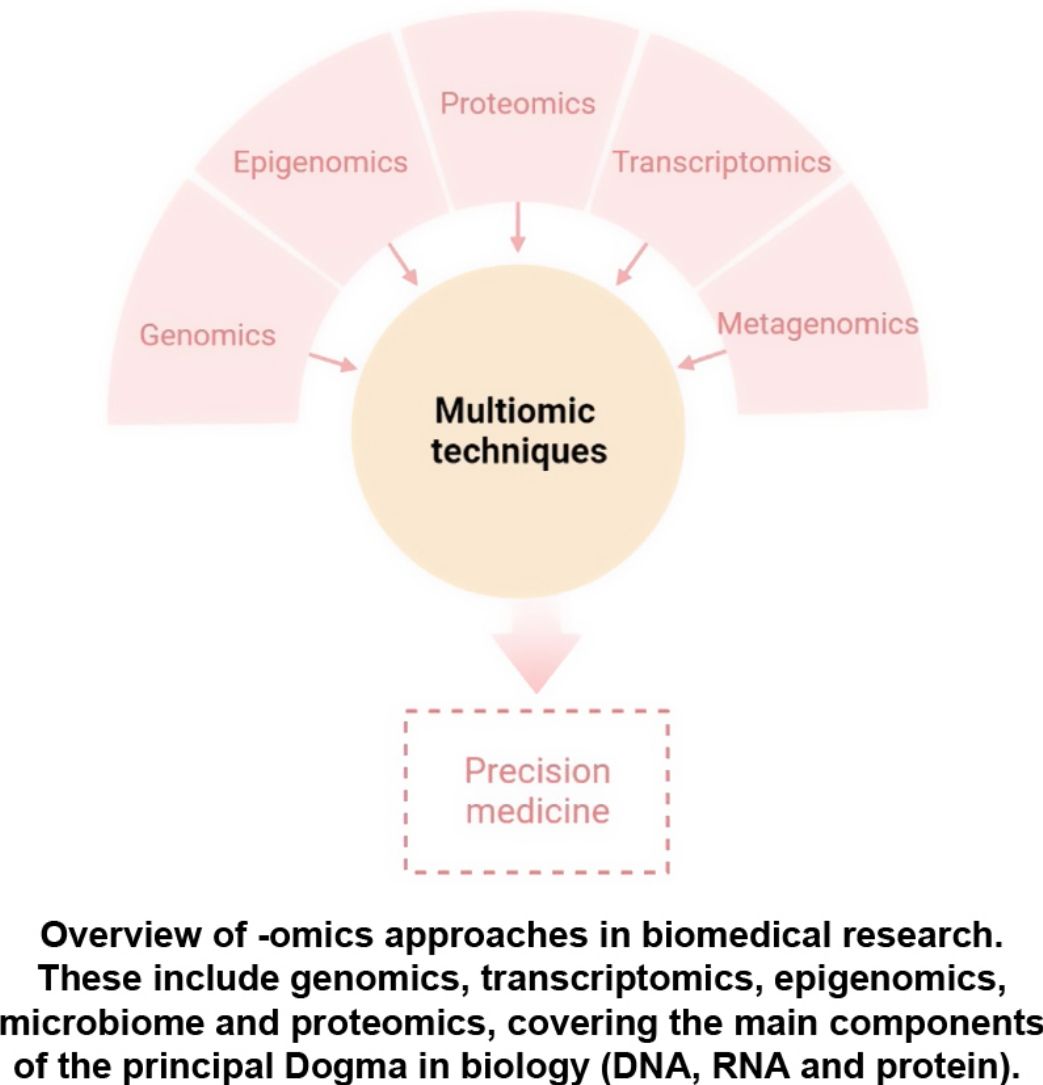
Atopic dermatitis (AD) has undergone, during the last decade, a revolution in pathogenic knowledge that has allowed the approach, for the first time in its history, of personalized therapeutic strategies. However, AD is a complex disease, very heterogeneous not only from the clinical point of view, but also with important variations associated with age, ethnic groups, and the presence or absence of atopic comorbidities and concomitant diseases. The large amount of knowledge generated has the risk of not being able to adequately characterize the broad spectrum of patients included under this spectrum if it is not properly integrated and can be interpreted by the clinical dermatologist.
The aim of this review is to provide clinical dermatologists with an overview of the nature and objectives of the main omics technologies (Fig. 1).
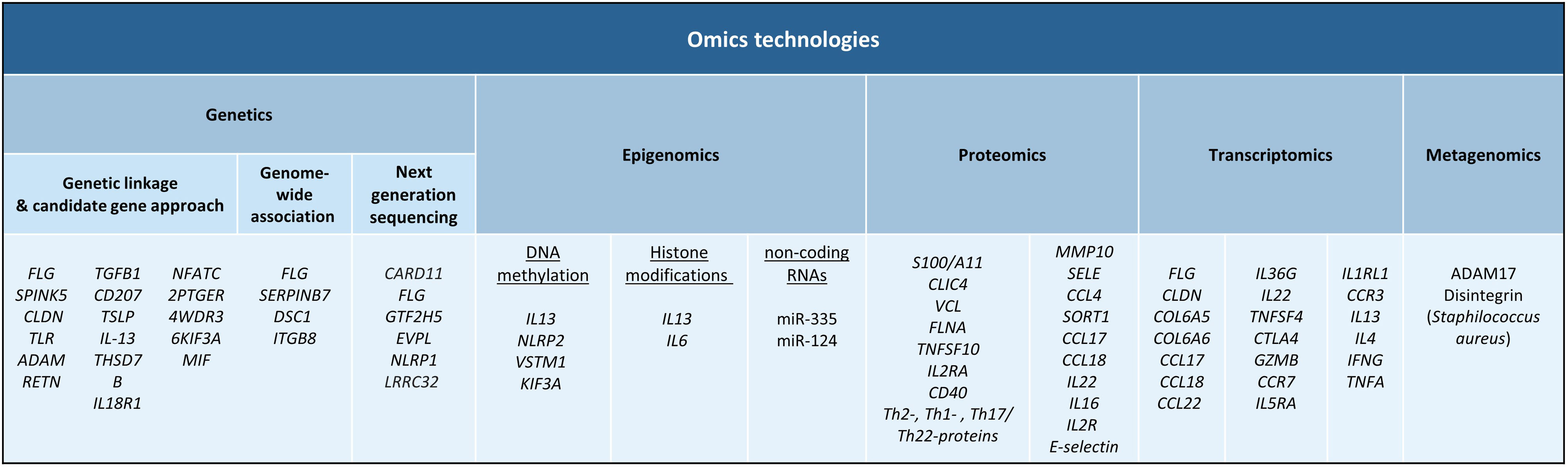
Atopic dermatitis (AD) is a complex immune-mediated inflammatory disease that has a strong genetic component.1 The heritability of AD has been estimated to be 70–80%,2,3 demonstrating the significant role that genetic factors play in the development of this chronic inflammatory disorder. In particular, the family history of AD has been described as the strongest disease risk factor. To date, genetic risk factors underlying AD have been identified using genetic linkage studies, candidate gene approaches, genome-wide association studies (GWAS) and, more recently, next generation sequencing (NGS) technologies have also contributed to better understand the genetic risk basis of AD.
Linkage studies and candidate gene approachesGenetic linkage studies are aimed at comparing the inheritance of genetic markers with the presence of a clinical trait to identify disease risk genes. In 2000, the first genome-wide linkage study in AD identified a major susceptibility locus on chromosome 3q21.4 The candidate gene approach, where variation in genes that are suspected to be involved in disease pathogenesis is compared between affected and healthy individuals, has been used by more than 30 studies to identify loci associated to the development and severity of AD.5FLG, the gene encoding for filaggrin, is one of the most well-known genes associated with both disease susceptibility and disease severity.1 Multiple genes supported by their functional implication in disease biology have been also studied as genetic determinants for AD risk.5 For example, loss-of-function mutations in the FLG gene have been originally found to be a major driver of AD development, and these mutations result in loss of filaggrin expression, a structural protein that binds keratin fibers in the epithelium.6,7 Also, studies conducted in the Asian population have identified a significant association between SPINK5 gene, which encodes for a serine protease inhibitor that participate in maintaining the skin barrier, and AD susceptibility.8 More than 25 genetic risk factors have been also detected through a candidate approach, including genes from the claudin (CLDN) and toll-like Receptor (TLR) families as well as ADAM, RETN, TGFB1, CD207, TSLP, IL-13, THSD7B, IL18R1, NFATC2, PTGER4, WDR36, KIF3A or MIF, among others.5 With the advent of high-throughput and next generation sequencing technologies, linkage and candidate gene analyses have been replaced by more powerful analytical strategies that allow a comprehensive analysis of genetic risk variation at the genome-wide scale.
Genome-wide association studies (GWAS)GWAS is a statistical approach that compares the allele frequency of millions of genetic variants between patients and healthy subjects to identify loci associated with clinical traits.9 To date, a total of seven GWAS have been conducted in AD.10–16 The summary statistics resulting from these GWASs conducted in populations of different ancestries have been further meta-analyzed.17–22 Altogether, these studies have identified more than 40 genes associated with AD risk. These include well-established risk genes such as FLG as well as other risk loci like SERPINB7, DSC1 and ITGB8, among others. Downstream analyses of GWAS findings have also revealed substantial overlap of AD genetic risk markers with enhancers characteristic of skin cells and immune cells like CD4+ T cells, providing evidence of novel biological pathways involved in AD pathophysiology.
In phenome-wide association studies, where genetic variation of interest is simultaneously tested for association with multiple clinical phenotypes, a loss-of-function variant in FLG has been found to be associated with different clinical phenotypes of AD like asthma, allergic rhinitis and food allergy.23 Genetic variation at ADAM33 metalloprotease and monocyte receptor CD14 genes has been also associated with allergic bronchitis and asthma, respectively.24
Next generation sequencingWhole exome sequencing (WES) technology, which allows a high-confidence identification of genetic variation from exonic regions, is being increasingly used to advance in the characterization of the genetic factors underlying AD.25–28 Of relevance, a single mutation in CARD11 has been found to cause potentially correctable cellular defects that lead to AD.29 Also, WES performed on AD patients from the African population has revealed rare variants in FLG and several other genes within the epidermal differentiation complex that are important for disease development, as well as nonsense and missense mutations in previously unreported genes like GTF2H5, EVPL or NLRP1.30 Further studies based also on next generation sequencing technologies have contributed to identifying low-frequency and rare missense mutations at LRRC32 gene in patients with AD,31 as well as additional mutations at FLG gene that are associated to AD development.32–34
Polygenic risk scoresFrom the clinical perspective, there is an urgent need to prevent the development of AD and its associated allergic comorbidities.35 In the last years, polygenic risk scores (PGSs) are being increasingly used to identify those individuals that are at high risk of AD and, therefore, more likely to benefit from an early therapeutic intervention.35–37 The PGS is a quantitative measure that estimates the genetic risk burden of an individual to develop a particular disease.38 This score is computed by aggregating the risk conferred by multiple small-effect variants that are distributed across the whole genome.39 Although only a few PGSs for AD have been developed so far,40 these genetic-based scores have already demonstrated a high power to predict the development of AD, reaching up to 85% of diagnostic accuracy. Like in cardiovascular diseases,41 integrating PGSs with established clinical risk factors could help physicians to detect those patients that are at the highest risk of AD and, consequently, to anticipate therapeutic interventions.
Mendelian randomizationDetermining causality in diseases like AD is key to develop better therapies and understand patient heterogeneity. Mendelian randomization (MR) is a statistical technique that uses genetic variants as instrumental variables to assess the causal relationship between an exposure and an outcome. With the increasing availability of GWAS, MR has gained popularity in recent years as a method to investigate potential causal relationships in observational studies. By using genetic variants as proxies for the risk factor of interest, MR minimizes the risk of confounding and reverse causation, and provides a more robust estimate of causality. MR has been applied to various research areas, including the study of the causes and consequences of AD. In particular, novel and valuable causal relationships between AD and epidemiological,42 clinical43 and molecular traits44 have been provided by MR.
EpigenomicsThe prevalence of AD has been increasing too rapidly to be accounted for by shifts in genetic variation. These changes are thought to be due to environmental factors, such as lifestyle, diet and pollution, mediated by epigenetic control of gene expression. The epigenome can be divided into three main groups: DNA methylation, histone modifications and non-coding RNAs.
DNA methylation occurs when DNA methyl transferases (DNMTs) catalyze the addition of a methyl group to the fifth carbon of cytosine resulting in 5-methylcytosine (5mC) and the presence of this methyl group usually correlates with transcription repression.45 Several studies have sought to investigate DNA methylation changes in AD with the objective of determining a mechanistic impact on pathogenic gene expression. One of the earlier studies that interrogated DNA methylation differences between AD and healthy controls found that although there were no DNA methylation changes in immune cell populations, there were striking DNA methylation alterations in AD lesional epidermis compared to healthy controls. These changes partly correlated with alterations in expression of genes that participated in keratinocyte differentiation and innate immune response.46 In a more recent study, the authors observed cell specific-DNA methylation in circulating T cells of AD patients.47 More specifically, skin homing CD4+ CLA+ T cells from AD patients were found to upregulate IL13 expression, a known Th2 AD marker, because of reduced DNA methylation upstream of that gene. Other studies identified altered DNA methylation in key pathogenic genes, and these include NLRP2 promoter hypermethylation in blood cells of pediatric AD patients,41,48 SNP-mediated demethylation of VSTM1 promoter in AD lesioned skin,49 and hypermethylation, thus downregulation, of the KIF3A gene that codes for a structural protein.50
Histone modifications are key epigenetic regulators that control chromatin structure and thereby control transcriptional events. The role of histone modifications has not been well-explored in AD. Nonetheless, alterations in histone modifications have been described in related inflammatory contexts, for example, one study described an increase in the transcriptionally active H3ac and H4ac histone marks in CD4+ T cells of asthma patients that correlated with increased IL13 expression.51 A more recent study showed that butyric acid isolated from Staphylococcus epidermidis, a known pathogenic bacterium in AD, functions as a histone deacetylase inhibitor and induced H3K9 acetylation in human keratinocytes that correlates with a decrease in IL6 production.52 Interestingly, microbiome derived butyrate has shown to provide protective functions in the intestinal tissue, indicating a highly contextual effect of metabolites in immune cell response.
Non-coding RNAs are functional RNAs that are not translated to proteins. Some of these RNAs, such as micro RNAs (miRNAs), directly control gene expression by mediating post-translational inhibition of mRNA transcripts by targeting them for degradation. In AD, one miRNA, miR-335, has been found to be downregulated in affected skin lesions compared to healthy controls, and its target SOX6 is aberrantly upregulated.53 Another key miRNA that has been described to be altered in AD is miR-124, which is downregulated in AD lesional skin compared to non-lesional skin. Authors found that miR-124 expression is essential for skin homeostasis by regulating p65 activity, and its downregulation in AD results in p65-mediated inflammation.54
ProteomicsIt has been well-accepted that although transcriptomic analysis can be very insightful, control of protein expression can be independent from gene expression. Hence, proteomics studies can be key to determining the mechanisms that drive pathogenesis of a disease. Conventional methods to study proteins include enzyme-linked immunosorbent assay (ELISA) and western blotting, and SDS-PAGE and 2D gel electrophoresis and related techniques are used for separation of complex protein samples.55 Subsequently, mass spectrometry (MS)-based methods have dominated the field of high throughput proteomics and have been extensively used to study numerous disease contexts. Since then, other high-plex methods to study proteomics have been developed, including antibody/antigen arrays,56 aptamer-based assays57 and proximity extension assay (PEA),58 and these methods have been key to understand disease mechanisms and to guide biomarker discovery.
Proteins that were initially found to be implicated in AD pathogenesis using conventional proteomics methods conducted in affected skin included functional proteins calcium-binding protein S100/A11 and CLIC4, and structural proteins vinculin and filamin A.59–61 Subsequent studies using liquid chromatography–mass spectrometry (LC–MS) revealed significant reductions in levels of proteins related to the generation of natural moisture and barrier function, suggesting that the continuous cycle of dry skin can predispose AD patients to infections.62,63
In a study by Pavel et al.,64 the authors observed elevated inflammatory protein markers in AD lesional skin that included proteins involved in immune cell activation (TNFSF10, IL2RA and CD40) and Th2-, Th1- and Th17/Th22-related proteins. Interestingly, these alterations were also seen non-lesional skin biopsies from AD patients. Furthermore, many changes that were detected at the protein level were not detected at the gene expression level, which supports the need for proteomics studies. Authors also investigated matched serum proteomics from the same patients and found that few proteomic changes could be detected in the serum. Amongst the detected alterations, the majority overlapped with what were detected in the skin, and these included proteins associated with Th1-, Th2- and Th17-related immune pathways.64
Other proteomics studies in serum not only demonstrated increased systemic inflammation mediators but increased cardiovascular risk (MMP10, SELE) and atherosclerosis (CCL4, SORT1) markers,65,66 indicating its relevance to study biomarkers that indicate comorbid inflammatory conditions.
Finally, proteomics approaches have been exploited to assess patient response to treatment. Thijs and colleagues analyzed cytokine levels in serum samples from patients before and after treatment with potent topical corticosteroids, and observed a decrease in several chemokines and cytokines, including CCL17, CCL18, IL22 and IL16, as well as IL2R and E-selectin.67 Another study showed that serum levels of IL16 positively correlated with disease severity and declined upon clinical improvement following topical treatment.68 A recent randomized clinical trial involving JAK/SYK inhibitor ASN002 showed that treated patients downregulated systemic inflammatory protein markers, as well as markers for atherosclerosis risk.69
These results indicate that AD is a true systemic inflammatory condition and that the circulating proteome is a direct reflection of local skin inflammation and should be exploited for biomarker discovery.
TranscriptomicsExtensive transcriptomics studies have been conducted in AD with the aim to decipher its physiopathology. It has been well-established that there are global changes in AD transcriptomic landscape at the site of inflammation, more specifically in the epidermis and dermis, mainly driven by activated keratinocytes and fibroblasts and infiltrating immune cells.
Firstly, several structural proteins are deregulated in AD, leading to major disruptions in skin barrier. Since then, aberrant expression of several other structural proteins has been also described, including proteins involved in tight junction formations, such as several members of the claudin family,70 and collagen production, such as COL6A5 and COL6A6.71 Secondly, apart from barrier genes, AD keratinocytes deregulate many genes in the lesional environment that perpetuate pathogenesis. It has been observed that AD keratinocytes upregulate proteases, such as kallikreins and serpins, and downregulate protease inhibitors.71 Furthermore, genes related to lipid metabolism and metabolic functions are also altered.72 Most importantly, these pathogenic keratinocytes upregulate a range of cytokines and chemokines, including CCL17, CCL18, CCL22 and IL36G, that play important roles in Th2- and Th22- and Th17-mediated inflammatory responses.70,71 Finally, major disturbances in skin immune cell populations have been observed in AD lesional skin transcriptomic studies. Deregulated genes include key mediators of T cell activation, such as IL22, TNFSF4 and CTLA4, cytotoxic activity, GZMB, and genes associated with T cell trafficking, including lymphoid organ homing receptor CCR7.70
Although skin transcriptomic data is highly informative regarding the general characterization of AD pathology, robust evidence suggests a systemic component that drives disease mechanisms. One study performed transcriptomic analysis in whole blood samples from pediatric AD patients and observed that eosinophil and Th2 markers, including IL5RA, IL1RL1, CCR3, IL13 and IL4, were upregulated while Th1 markers, such as IFNG and TNFA, were downregulated.73 A more recent study corroborated these findings in adult AD patients.74 Furthermore, authors were able to identify two transcriptionally distinct AD endotypes, annotated as eosinophil-high and eosinophil-low, characterized by differential expression of eosinophil-related signature. The eosinophil-high endotype displayed more pronounced global dysregulation and continued disturbance of NK cell function despite clinical improvement following treatment. More interestingly, the eosinophil-low endotype corresponds to a higher proportion of super responders to dupilumab, an anti-IL4RA therapy.74
Overall, major transcriptomic alterations drive local skin inflammation in AD lesions. However, understanding systemic transcriptomic changes in circulating immune cells appears to be key in both the search for new therapeutic treatment and in determining response to treatment.
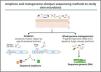
Single-cell omicsThe cellular content of human skin has been traditionally studied using strategies combining fluorescence-activated cell sorting (FACS) and immunohistochemistry (IHC) on enzymatically digested tissue. With the recent implementation of single-cell omics approaches researchers can carry out a comprehensive molecular and functional characterization of skin cells to understand the molecular bases of AD.75 Among all the available single cell technologies, the droplet microfluidics-based protocol commercialized by 10X Genomics is the most popular to study the transcriptome of single cells (scRNA-Seq) in dermatological research. This technique is based in the encapsulation of cells into microdroplets with cellular barcodes and unique molecular identifiers (Fig. 2).
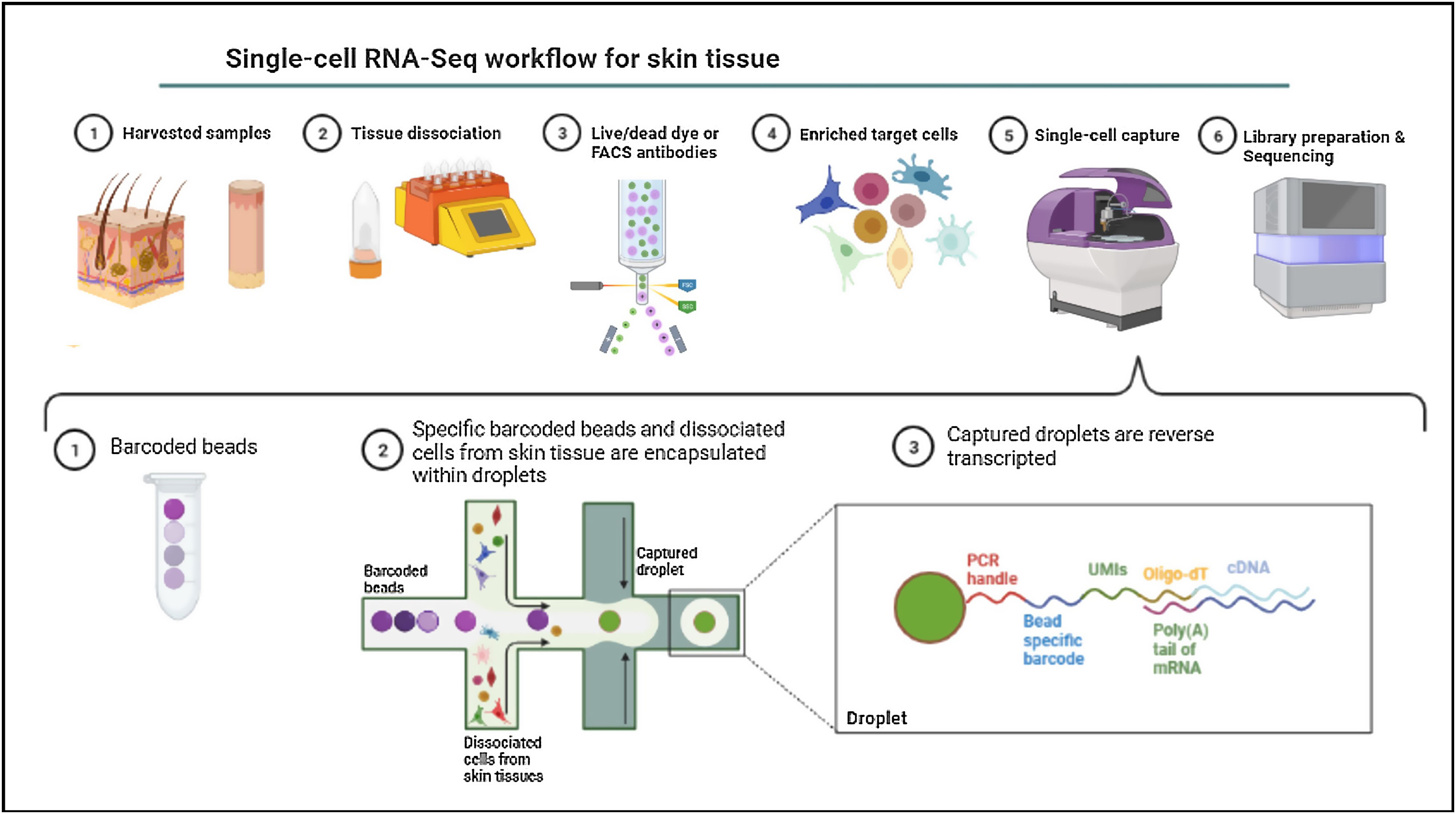
The isolation of single cells from skin biopsies requires an initial step of mechanical or enzymatic treatment, which can be quite challenging due to the structural complexity and heterogeneity of skin. Thus, the dissociation approach used for the obtention of isolated cells needs to be adapted to the properties of the skin layer of interest.76
The comparison of skin from AD, and healthy volunteers using scRNA-Seq has revealed the expansion of ILC2, type2/type 22 T cells, inflammatory DCs and fibroblasts expressing pro-inflammatory cytokines, such as CCL2 and CCL19, in lesional AD samples.77–79 In pre-clinical assays, it has been shown that the expression of CCL11 in a specific population of fibroblasts from AD subjects contributes to the pathogenesis through IKKB/NF-kB signaling.80 The study of the transcriptomic and proteomic profiles of skin cells from spontaneously resolved AD, chronic AD, and healthy donors revealed great regulatory differences in both immune and non-immune cells among the tested samples.81 It was also found that melanocytes rather than immune cells, show the highest number of differentially expressed genes between spontaneously healed AD and controls. This unexpected immunological role of this cell type in AD clearly demonstrates the power of single cell approaches to generate new pathogenic proposals of the disease.
A recent study involving different skin conditions reported the transcriptional dysregulation of skin-resident memory T cells in AD and psoriasis. This T cell program would likely be a key contributor to disease recurrency.82 Regulatory abnormalities involving T helper markers in ILC and CD8+ cytotoxic T cells were exclusively observed in AD. The characterization of these transcriptional changes allowed for the definition of major classes of disease classes associated with therapy response.
In summary, the studies of cell composition and transcriptional traits of AD carried out in the last four years, have corroborated the role of immune cells and fibroblasts in AD development, and the relevance of melanocytes in the maintenance of regulatory microenvironment in AD remission.
MetagenomicsThe human skin is colonized during the postnatal period by microorganisms that prevent the invasion of external pathogens. This collection of commensals that include bacteria, fungi, and viruses, is known as skin microbiota. Crosstalk between skin microbiota and host immune system is necessary to trigger homeostatic activation of the innate and adaptive immune systems.83
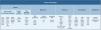
In the last decade complex microbial communities, such as the human skin microbiota, have been explored using metagenomics, which circumvents the bias imposed by traditional bacterial culture in artificial growth conditions. Two different metagenomic approaches have been developed to determine and quantify the composition of microbial communities: amplicon sequencing and whole metagenome sequencing (Fig. 3).
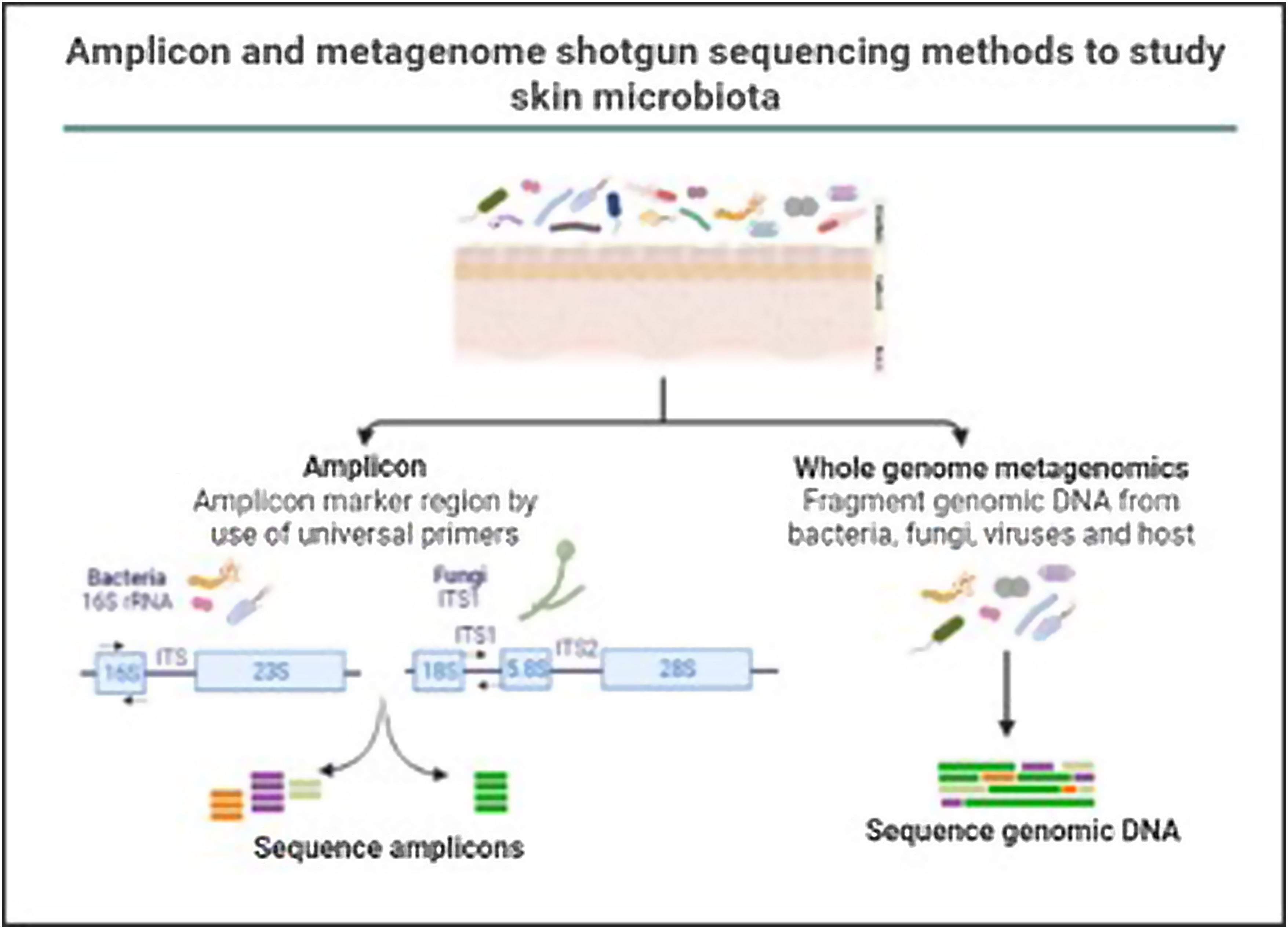
Amplicon and metagenome shotgun sequencing methods to study skin microbiota. Amplicon-based strategy relies on conserved and variable fragments of bacterial 16S rRNA gene, and fungi ITS1 region. With whole metagenome sequencing all genomic DNA present in a sample is fragmented and sequenced.
The use of these metagenomics approaches has been key to characterize the imbalance of the microbial composition in AD patients, when compared to non-AD subjects.84 The colonization of Staphylococcus aureus, a pathogenic bacterium that causes skin infections, has been widely reported in AD skin. The first longitudinal study of clinical samples from pediatric AD using 16S rRNA sequencing showed great compositional differences in the skin microbiota of AD patients versus controls (Kong et al., 2012). An association between bacterial diversity and AD treatment, as well as a correlation between S. aureus and S. epidermis and disease flares, were reported too. Later studies performed with shotgun metagenomics corroborated these findings.85 Although the colonization of S. aureus in AD skin is recurrently observed, the contribution of this pathogen to the initiation of the disease has not been disentangled yet.
The study of genetically modified mice has made it possible to explore the role of the skin microbiota in the initiation of inflammatory skin diseases. Eczematous dermatitis has been recapitulated in mice deficient in disintegrins and ADAM17 genes, which induced skin dysbiosis.86 The skin of these mice was enriched in S. aureus, Corynebacterium mastiditis and Corynebacterium bovis. The treatment with antibiotics targeting these pathogens reversed the atopic skin phenotype and eliminated inflammation, which demonstrated a likely causal link between skin microbiome alterations and dermatitis. Thus, novel therapies targeting S. aureus and recurrent commensals enriched in AD skin are needed to replace the broad-spectrum antimicrobials that are currently used in these patients.
In 2016, the skin microbiome of samples from volunteers with AD, healthy donors (HDs), and non-AD reported atopy (AD-susceptible subjects) was profiled, using both 16S rRNA sequencing and shotgun whole metagenome sequencing.87 Three different skin sampling methods were evaluated (skin swabbing, cup scrub, and tape collection), concluding that the combination of tape sampling and whole metagenome sequencing is an easy-to-use and robust skin microbiome analysis protocol. They observed that Streptococcus and Gemella genera were more abundant in AD than in healthy donors, whereas Dermacoccus, Deinococcus, and Methylobacterium were depleted in AD. At a functional level, the microbiome of AD subjects seemed to be primed to produce an excess of ammonia, providing an explanation for the high pH levels that are typically observed during atopic dermatitis flares. In addition, a higher proportion of eukaryotes was observed in healthy individuals than in AD, as well as a depletion of Malasseziaceae family in AD-susceptible individuals. Different members of the Malassezia genus, which are commensal fungus associated with AD, have been shown to induce inflammation and exacerbate AD symptoms.88
Interestingly, reductions of the microbial diversity and gammaproteobacteria abundance observed in skin from AD and controls have been associated with lower environmental biodiversity in the surroundings of skin donors’ homes.89 Gammaproteobacteria, a class associated with health in human skin,90 has been found to positively correlate to IL-10 levels in blood, a key anti-inflammatory cytokine involved in immunologic tolerance.
In a recent study, the skin microbiome of more than 822 Chinese and 538 American samples were collected and profiled with shotgun metagenomics, resulting in the integrated human skin microbial gene catalog (iHSMGC).91 This catalog of bacterial species represents a highly valuable reference tool to characterize AD-associated skin dysbiosis, allowing for the exploration of the AD resistome composition, as well as the potential function of the skin commensals in this condition.
ConclusionAny of the described molecular-based approaches provide a large amount of new and interesting information regarding AD. However, only a holistic, clinically driven vision that allows the integration of all the “omics” will make possible to conclude all this available information in a personalized medicine (Fig. 4).
In this objective, it is essential to join the efforts of clinicians and specialists in all these techniques to obtain the best clinical picture – and the more realistic – of the patients we see in our offices, the most accurate samples at the right time, and the most innovative techniques to be able to lead the qualitative leap that the possibilities in “omics” represent. The SSAD study, a prospective multicenter study that will bring together the work of more than 30 Spanish centers specialized in AD, has been born and designed under this premise.
FundingThis work and the article processing charges were funded by IMIDomics, FIS grant (PI21/01139) and EADV project (PPRC 2021-22). The sponsors had no role in the design, execution, interpretation, or writing of the work.
Conflict of interestsSM is co-founder of IMIDomics, Inc. AJ is Chief Data Scientist of IMIDomics, Inc. The remaining authors declare that they have no conflict of interest.
The authors thank all the physicians and health professionals who contributed in the Spanish IMID Consortium as well as the AD participating Clinical Centers.


