



Sturge-Weber syndrome is a sporadic congenital neurocutaneous disorder caused by a somatic activating mutation in GNAQ; it affects 1 in every 20,000 to 50,000 newborns. It is characterized by a facial Port-wine stain, leptomeningeal angiomatosis, and glaucoma. Seizures are the most common neurological manifestation and typically present in the first months of life. Glaucoma may be present at birth or develop later. Neuroimaging studies show leptomeningeal angiomatosis, supporting diagnosis. Standard treatment for Sturge-Weber syndrome includes laser treatment for the Port-wine stain, anticonvulsants, and medical or surgical treatment for the glaucoma. Prognosis depends on the extent of leptomeningeal involvement and the severity of the glaucoma.
El síndrome de Sturge Weber es un trastorno neurocutáneo congénito, esporádico causado por una mutación somática activadora en el gen GNAQ, con una incidencia de uno de cada 20,000-50,000 nacidos. Se caracteriza por la presencia de una mancha en vino de Oporto facial, angiomatosis leptomeníngea y glaucoma. La manifestación neurológica más común son las convulsiones, que suelen comenzar en los primeros meses de vida. El glaucoma puede estar presente desde el nacimiento o desarrollarse posteriormente. Los estudios de neuroimagen permiten visualizar la angiomatosis leptomeníngea, ayudando al diagnóstico del síndrome de Sturge Weber. El tratamiento estándar incluye láser para la mancha en vino de Oporto facial, anticonvulsivantes y tratamiento médico o quirúrgico del glaucoma. El pronóstico de la enfermedad dependerá de la extensión de la malformación leptomeníngea y del grado de afectación ocular.
Sturge Weber syndrome (SWS) is a neurocutaneous disorder that is associated with facial capillary malformation (Port wine stain [PWS]), glaucoma, and leptomeningeal angioma in its complete form. Its incidence is estimated to be 1 per 20 000-50 000 live births.1 Some authors use the term SWS to apply to incomplete forms in which only 2 of the aforementioned manifestations are present. Some, for example, classify encephalotrigeminal angiomatosis according to the Roach scale in 3 types: type i with facial PWS and leptomeningeal angiomatosis, with or without associated glaucoma, corresponding to classical SWS; type ii, which is more common, with facial PWS and no leptomeningeal involvement, with or without presence of glaucoma; and type iii, which is the least frequent form, with presence only of leptomeningeal angiomatosis.2
EmbryologyCapillary malformation (PWS) and leptomeningeal angiomatosis may result from failure of the primitive cephalic venous plexus to regress. In early stages of development, the primitive venous system is divided into an external portion, which feeds and drains the facial skin and scalp, a middle portion, which feeds the meninges, and a deep portion, which feeds and drains the brain.3 It has been suggested that in this stage, the embryological proximity of the ectoderm, destined to form the upper portion of facial skin, to the portion of the neural tube that will form the parieto-occipital area of the brain, may explain the association between facial PWS and leptomeningeal angioma.4,5
Genetics and PathogenesisFor years, it was hypothesized that the cause of SWS might be a somatic mutation, and this would explain its sporadic appearance and the patchy distribution of abnormal blood vessels.4,6
In 2013, Shirley et al.7 detected an activating somatic mutation in the GNAQ gene, specifically substitution of a single nucleotide (c.548G→A, p.Arg183Gln) in 23 samples of affected tissue from 26 patients with SWS (88%), and in 12 out of 13 samples from patients with nonsyndromic PWS (92%). The prevalence of the mutation in involved tissue was between 1% and 18%, confirming its mosaic pattern.7 Other authors have confirmed that 80% of the patients with SWS have this somatic mutation in brain tissue.8
The extent of PWS and leptomeningeal and ocular involvement depend on the moment in which the mutation occurs and the cell affected.8
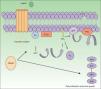
The GNAQ gene is located on chromosome 9 and is comprised of 7 exons that cover a region of 310 993 nucleotides. Its gene product is one of the 4 members of the Gq-alfa family, a class of G proteins that participate as modulators and translators in different transmembrane signaling systems. Somatic mutations in GNAQ, other than those detected in capillary malformations, have been reported in melanocytic neoplasms, uveal melanoma, congenital hemangiomas (RICH and NICH), and phakomatosis pigmentovascularis.9,10 Specifically, GNAQ and GNA11 mutations have been detected in phakomatosis pigmentovascularis, and the same mutation has been detected in the pigmentary and vascular component in the cases studied.11 These mutations increase proliferation and inhibit apoptosis by increasing signaling through RAS effector pathways (Fig. 1).
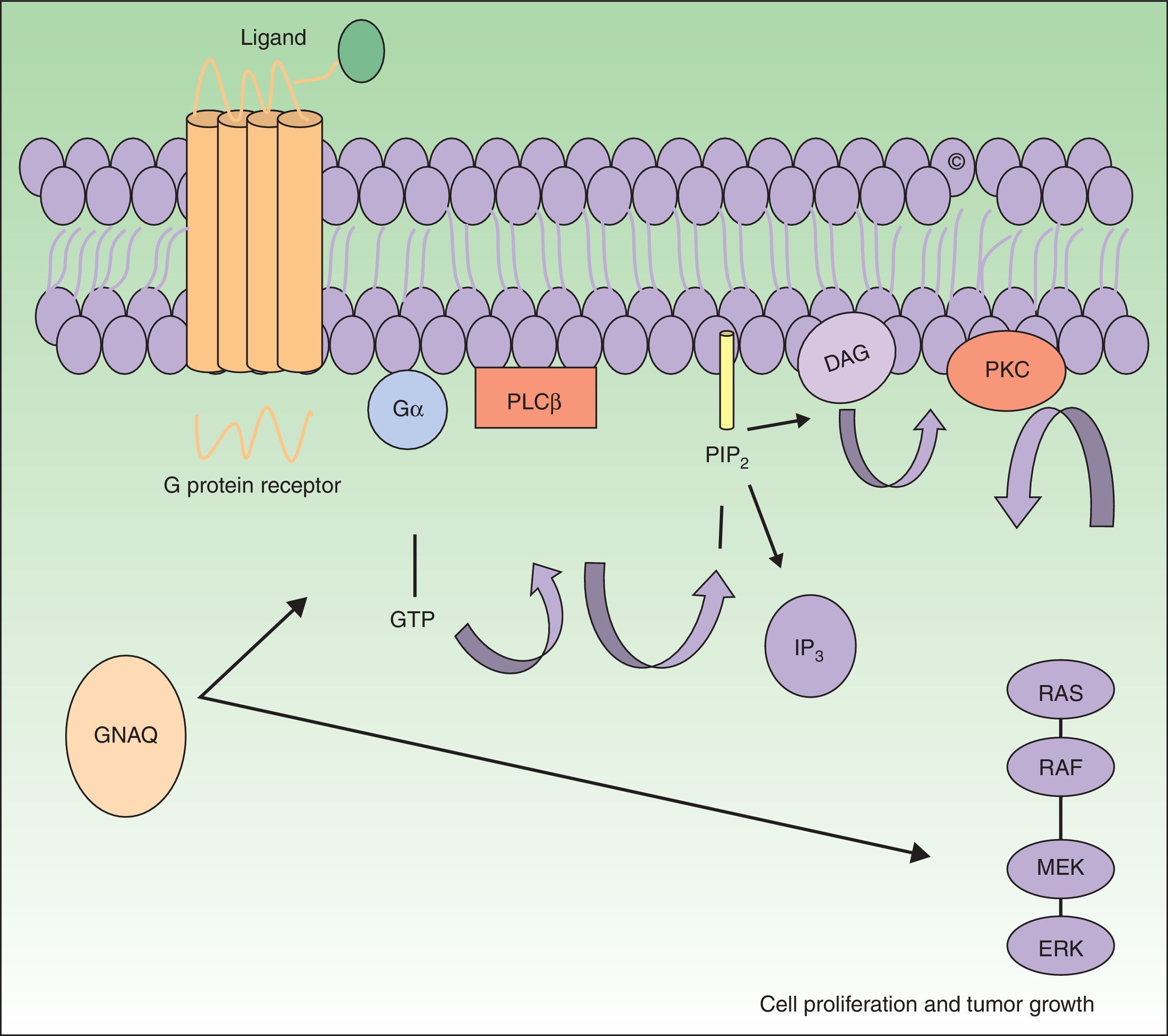
GNAQ is implicated in cell growth by signal transmission through cell membrane receptors via MAP kinases. An activating mutation therefore increases signaling via this pathway, and this may lead to the capillary malformations observed in Sturge Weber syndrome. Abbreviations: DAG, 1,2 diacylglycerol; Gα, G protein α; IP3, 1,4,5 triphosphate; PIP2, phosphatidylinositol 4,5-biphosphate; PKC, protein kinase C; PLCβ, phosphorylase C.
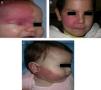
Capillary or venular malformation, also denoted as PWS or nevus flammeus, is the characteristic cutaneous manifestation in SWS.12 This lesion is already present at birth, of variable size, normally on one side though it may be bilateral, and of a color ranging from pale pink to purple. PWS can be confused with salmon patches (nevus simplex). Salmon patches are capillary malformations that present as a poorly delimited rose-colored macules, usually located in the central area of the forehead, philtrum, upper eyelids, vertex, and neck.13 The lateralized localization of PWS, its more intense color, and its well-defined borders help differentiate this lesion from salmon patches.
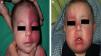
The risk of leptomeningeal or ocular involvement when facial capillary malformation (PWS) is present depends on the extent of the PSW and its site. Malformations that affect the frontal region have a higher risk than those that affect the lower part of the face.14 To date, studies have stratified risk assuming that facial PWS are distributed according to the areas of sensory innervation of the 3 trigeminal nerve branches: the frontal branch (V1), the maxillary branch (V2), and the mandibular branch (V3)15–18 (Figs. 2A-C). All studies agree that lesions with a highest risk of involvement are those affecting the V1 branch, the upper eyelid, or those that are bilateral and extensive (Fig. 3A and B). Thus, the incidence of leptomeningeal involvement or glaucoma in patients with capillary malformations with V1 involvement is between 8% and 15%, whereas if the involvement is bilateral or affects several dermatomes, the incidence is 28%.17 The risk that PWS with exclusively V2 or V3 involvement are associated with leptomeningeal involvement or glaucoma is almost negligible, according to most authors.17,18 However, in some studies, V2 involvement is considered to have a risk of 2%.17,18 This discrepancy may be because some authors have considered the lower eyelid as belonging to V2 whereas others consider that it belongs to V1.17,18
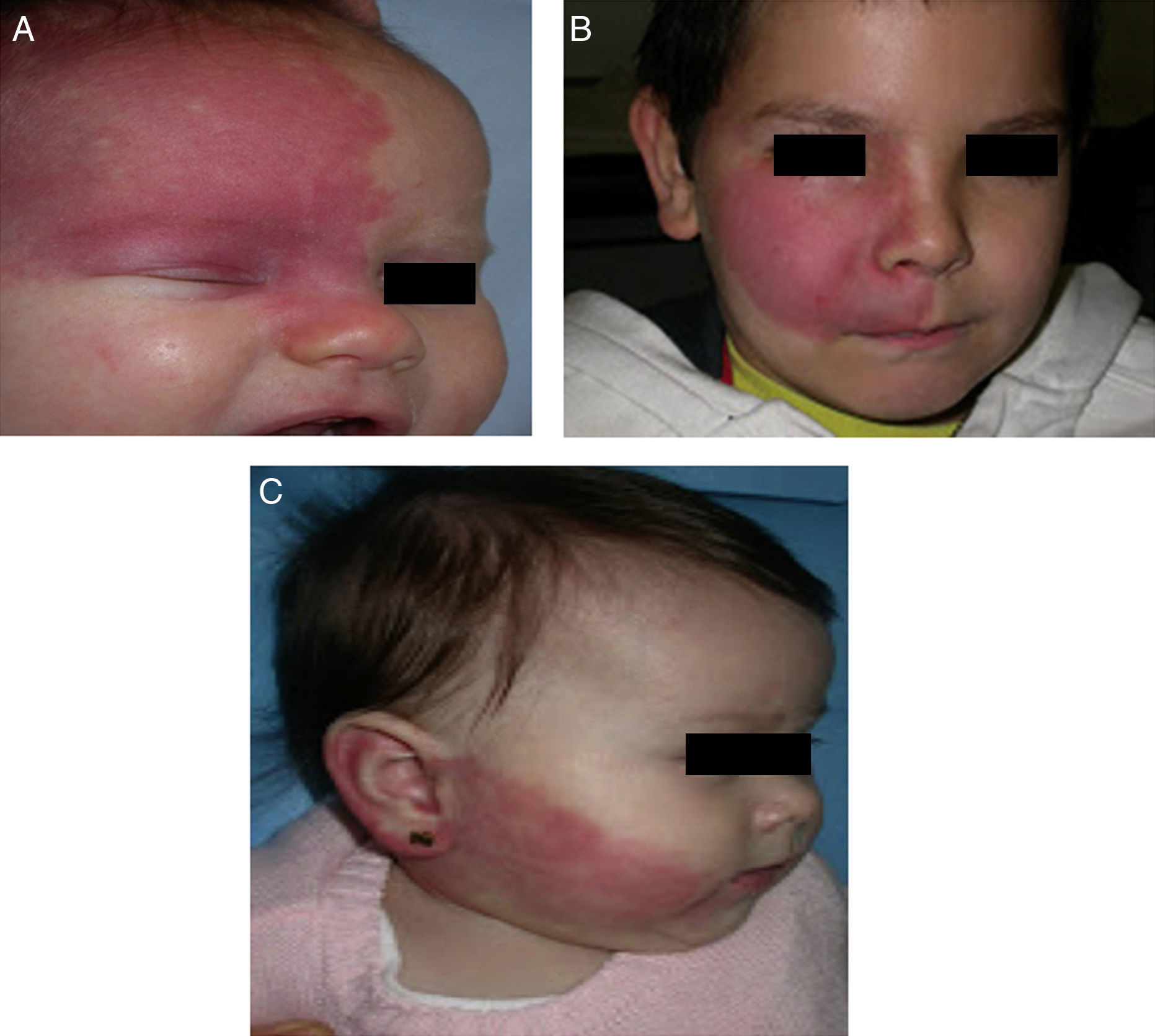
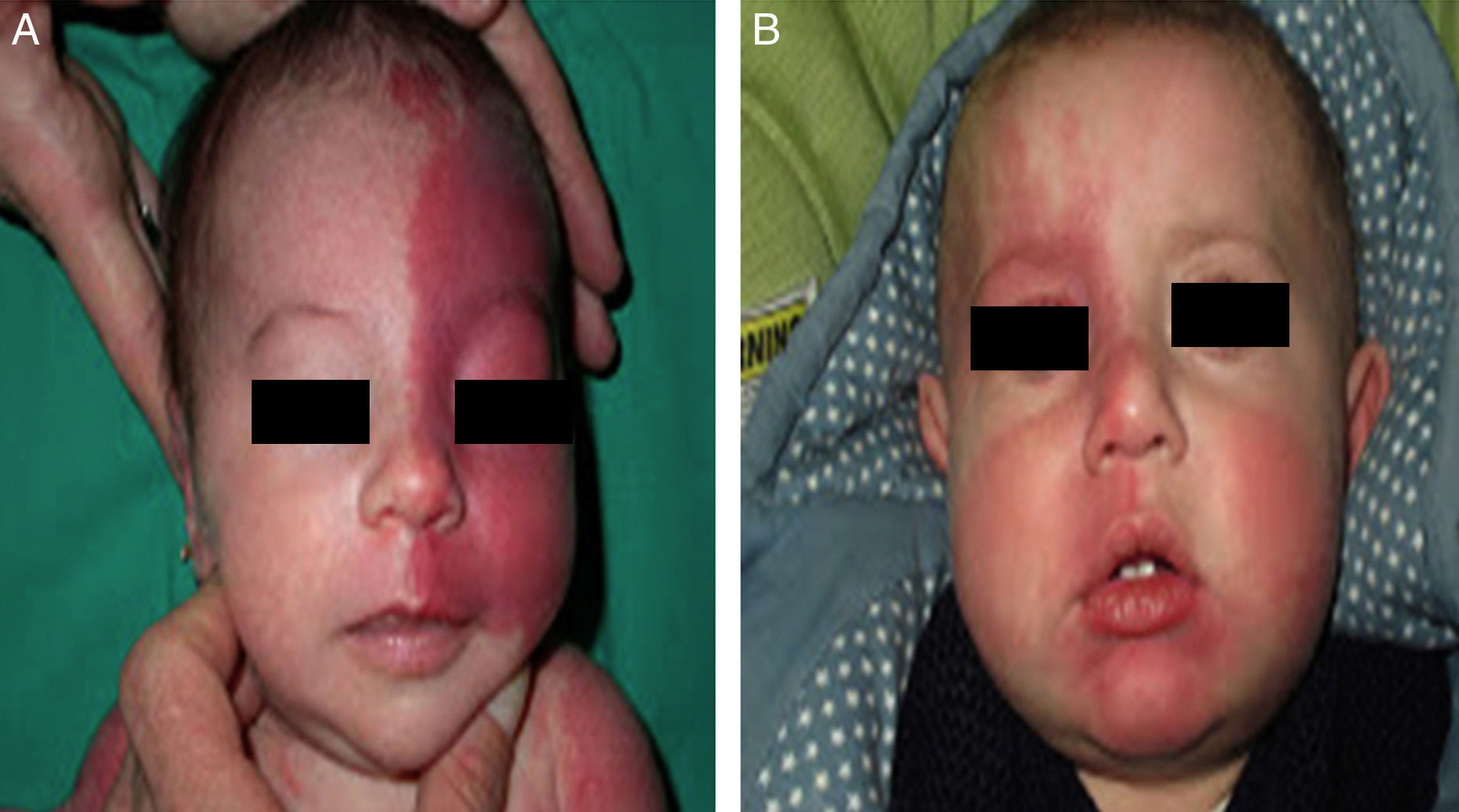
This distribution of PWS according to the sensory trigeminal nerve branches has been widely debated. Waelchli et al.,13 after reviewing the distribution of facial PWS in 192 children, arrived at the conclusion that the lesions do not follow the sensory areas of the trigeminal nerve but rather follow 8 more or less repetitive areas of distribution. According to these authors, the area of the forehead between the midline of the forehead and a line that joins the outer canthus of the eye and the top of the ear, including the upper eyelids, is the only area with a risk of association. All patients with abnormalities in magnetic resonance imaging (MRI), seizures, or glaucoma, had involvement of this frontal area. Of note, though, is that this retrospective study only performed MRI on 4 patients with PWS outside this area. The frontal area mimics the distribution, in the area of the optic vesicle, of the frontonasal prominence and skin, which are derived from the frontal placode. The placodes develop their own vasculature, and so the authors postulate that facial PWS imitate the embryological vasculature and do not follow the sensory nerve distribution of the face. The frontal placode develops its vasculature from the prosencephalon and anterior mesencephalon, structures that will give rise to the brain and optical vesicle, thus explaining why the PWS in this area derived from the frontal placode are those with a risk of SWS.13,19
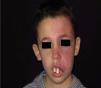
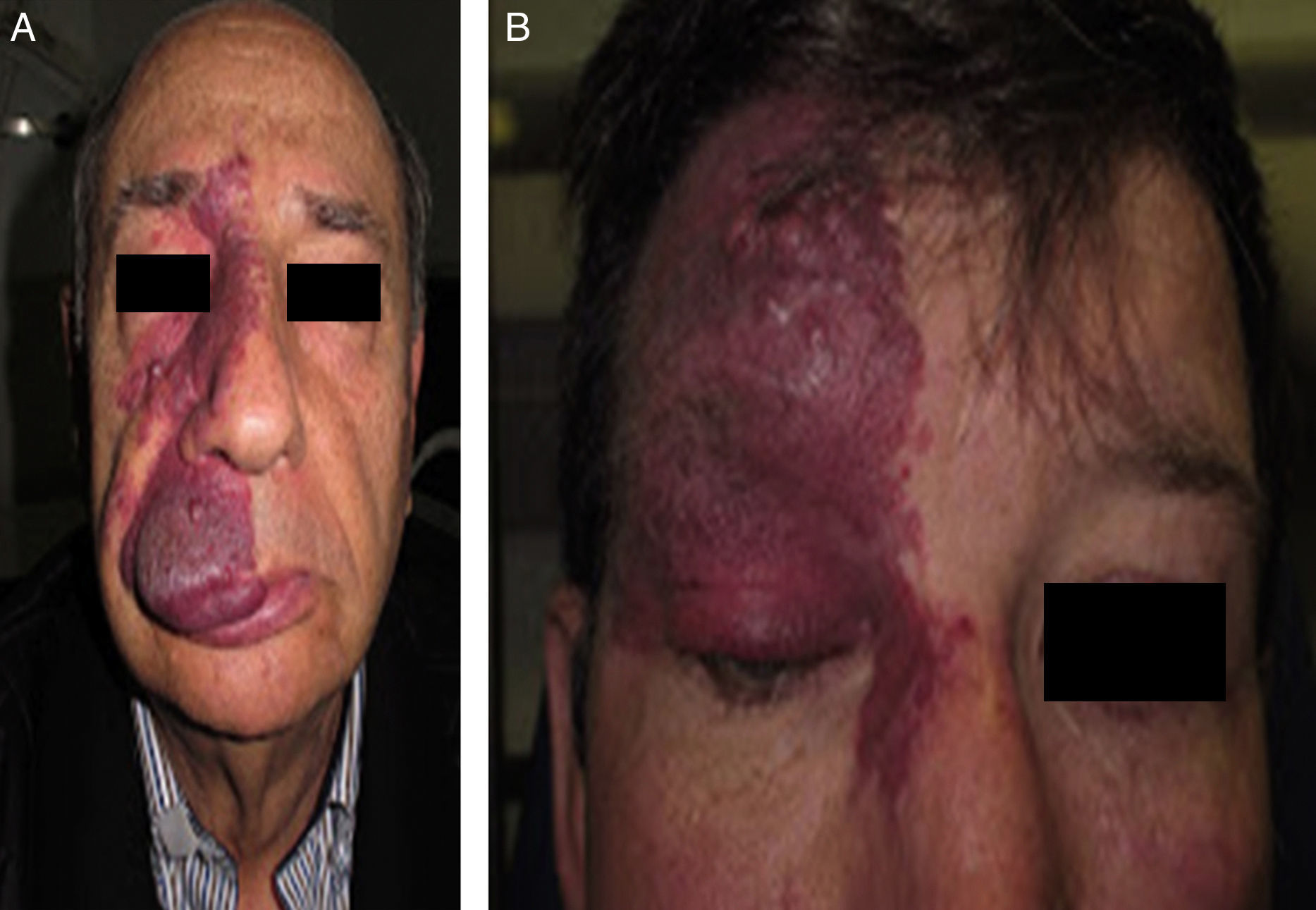
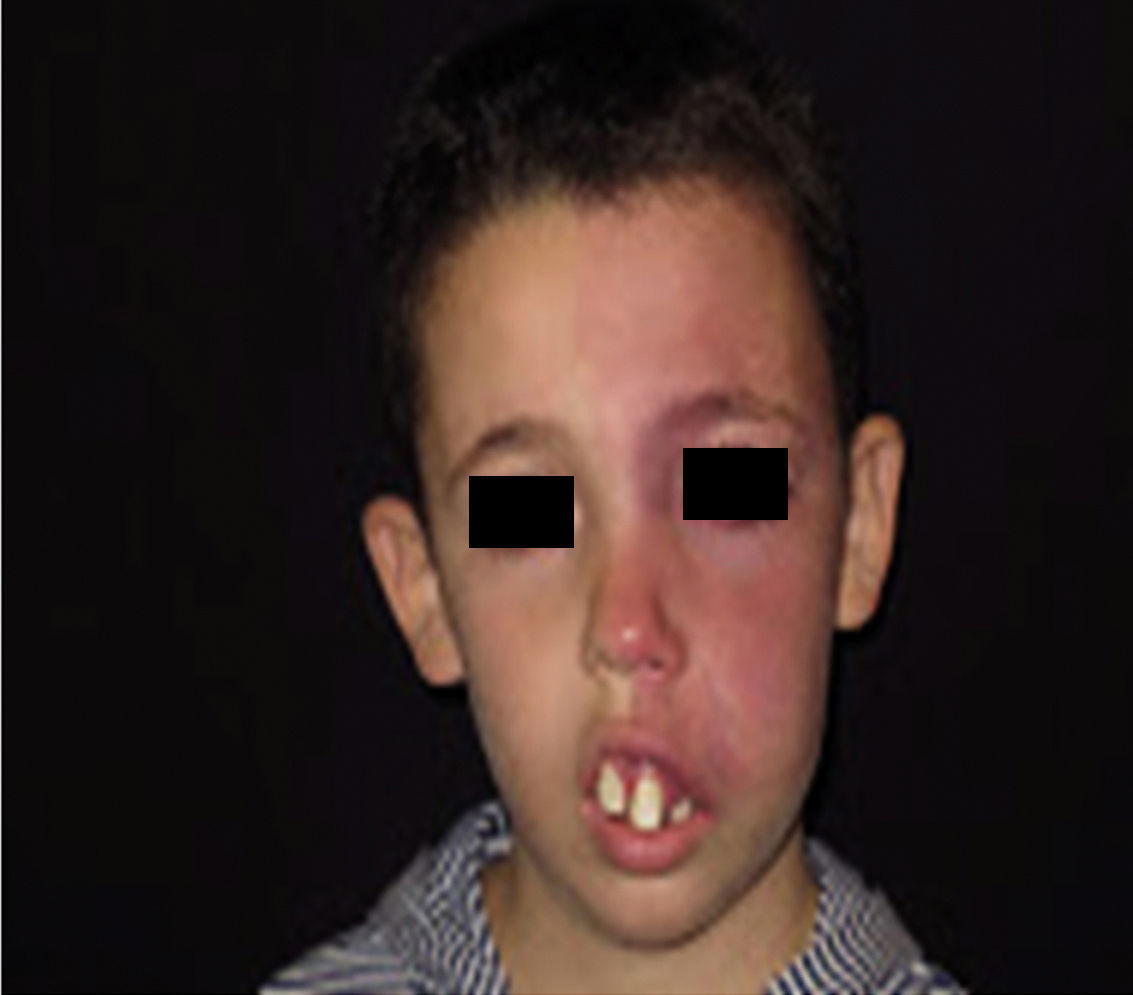
Facial PWS develop soft tissue hypertrophy in 60% of cases, bone hypertrophy in 13.8%, and formation of proliferative nodules or progressive ectasia in 43.8% (Fig. 4A and B). The mean age of onset of these changes is 9 years and the maxillary branch (V2) is the one which most frequently presents hypertrophy.20,21 Poor occlusion and greater dental exposition may result from excessive growth of the maxilla and jaw, leading to significant facial deformation22 (Fig. 5).
One of the manifestations of SWS is capillary-venous leptomeningeal malformation. Intracranial angiomatosis is usually ipsilateral to PWS, although it can be bilateral. It generally involves the occipital and occipitoparietal lobe and, at times, the entire hemisphere.1
Histologically, the cerebral vascular malformation consists of tortuous vascular structures and abnormalities in the thickened leptomeninges. Abnormalities are also observed in the deep draining veins, which are abnormally dilated. The underlying brain tissue may be atrophic and show neuronal loss, astrocytosis, cortical dysgenesis, and calcifications distributed perivascularly or in the cerebral cortex. It is thought that these are dystrophic calcifications caused by hypoxia. The leptomeningeal vessels of patients with SWS show overexpression of fibronectin and vascular endothelial growth factor (VEGF) as well as increased endothelial proliferation and apoptosis.4,23
The main clinical manifestations of leptomeningeal angiomatosis are seizures (75% to 90%), slowly progressive hemiparesis (25% to 60%), migraine-like vascular headaches (30% to 45%), delayed neuropsychological development (50% to 60%), episodes similar to cerebrovascular events, with acute transient hemiplegia, visual field defects, and behavioral problems.1,3 The seizures present in patients with SWS are the result of cortical irritation caused by cerebral vascular malformation through mechanisms of hypoxia, ischemia, and gliosis.24 Seizures are usually complex focal seizures or partial seizures with secondary generalization.23 Seizures first occur during the first year of life in 75% of patients and before the second birthday in 90%.25 Fever may trigger seizures in SWS. Patients without PWS but with leptomeningeal angiomatosis may present seizures during childhood or even as adults.14 When seizures first present before 2 years of age, they are more difficult to control and there is a greater risk of mental retardation.26,27 Progressive neurological deterioration in children with SWS is of ischemic origin, caused by altered cerebral perfusion and associated with elevated metabolic demand arising from prolonged seizure activity.4
Ocular EffectsOcular vascular malformation consists of dilated tortuous venous vessels that may involve the conjunctiva, episclera, retina and/or choroid, leading to optical atrophy and blindness.23
Glaucoma is one of the most frequent ocular manifestations in SWS, affecting approximately 30% to 70% of patients.25,28,29 It is usually unilateral or ipsilateral to PWS, but this is not always the case.1 Glaucoma in SWS is thought to be caused either by increased episcleral venous pressure, a pathophysiological mechanism supported by the presence of blood within the Schlemm canal, or by abnormalities in the anterior chamber which interfere with normal outflow of the aqueous humor, resulting in increased outlet resistance.23,30 Glaucoma can be congenital or develop after birth. In 60% of the early forms, glaucoma is caused by abnormalities in the angle of the anterior chamber, whereas the cause of glaucoma in 40% of young people and young adults is elevated episcleral venous pressure.30
Choroidal hemangioma is present in 40% to 50% of patients with SWS, and can be circumscribed and/or diffuse. At times, the vascular choroidal lesion takes on a pathognomonic character, such as a reddish hue, which has been described as tomato catsup color.24 The choroid normally remains unchanged during childhood; however, in adolescents and adults, it may become notably thickened.30
Other ocular abnormalities have been reported, although less frequently, for example, heterochromia iridium, detached retina, strabismus, homonymous hemianopsia, phakomatosis pigmentovascularis, iris and choroidal neovascularization, and lens luxation.31,32
Endocrine ManifestationsPatients with SWS may have hypothalamic-pituitary disorder, with growth hormone deficiency and central hypothyroidism.33–35
Additional StudiesHead X-RayAlthough plain X-ray is not a technique of choice, it is possible to observe the classical gyriform cortical calcifications also known as railroad track appearance, which affect the intimal layer of the meningeal arteries. These are adjacent to the leptomeningeal angioma, mainly in the parietal and occipital region.26,36 Calcifications usually affect children aged 2 years, and so they are considered a late-onset finding.37
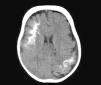
Computed TomographyBrain tomography is a technique used frequently in the emergency room to assess children who present with hemiparesis or seizure. The characteristic findings are presence of reduced parenchymal volume, enlarged ventricle, and enlarged choroid plexus. From 1 year of age, computed tomography can detect calcifications (Fig. 6), which would not normally be visible in a plain X-ray. Computed tomography is also more sensitive for detecting calcifications than MRI.14,37
Brain Magnetic Resonance Imaging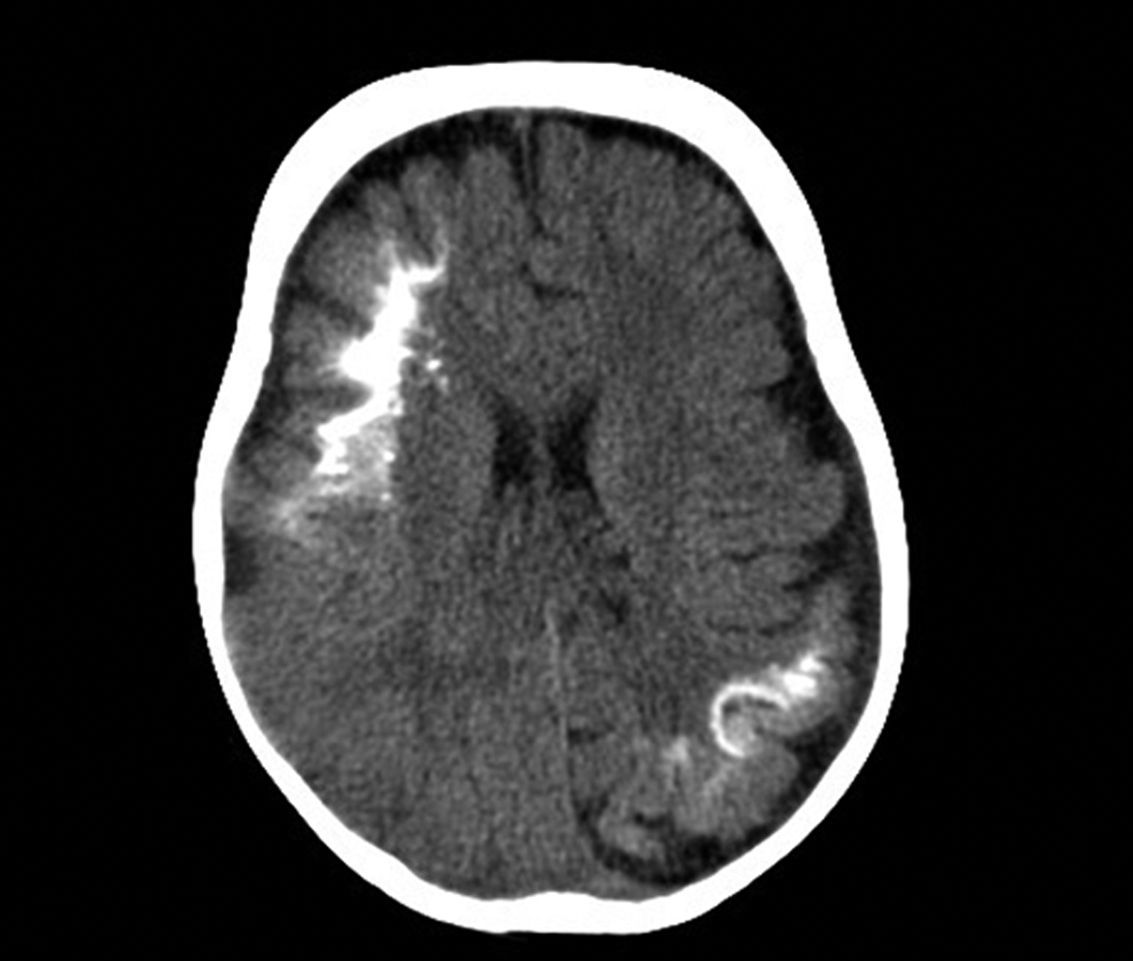
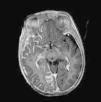
Brain MRI with gadolinium enhancement is the imaging technique of choice for diagnosis of SWS.28 With this technique, it is possible to visualize the leptomeningeal vascular malformation that confirms diagnosis of SWS (Fig. 7). Calcifications in MRI can also be detected in T2 sequences as images of cortical and juxtacortical hypointense signal. MRI can also detect abnormal venous drainage, reduced brain volume, enlarged ipsilateral choroid plexus, prominence of subependymal and medullary veins, loss of volume of the affected brain hemisphere, and accelerated myelinization underlying the leptomeningeal angioma.14,24,37 These changes are more evident after 1 year of age.38
Electroencephalogram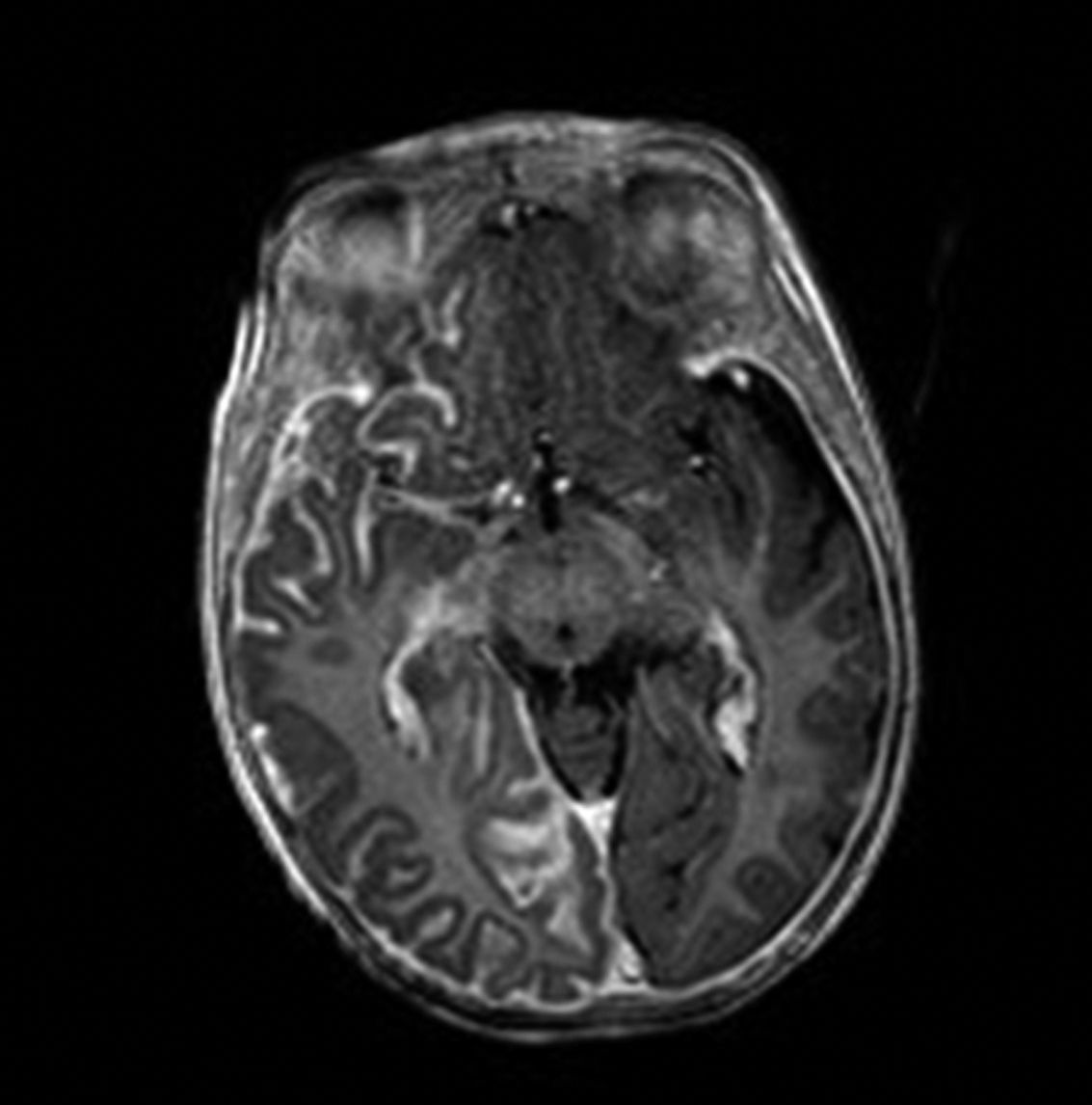
The characteristic electroencephalogram (EEG) of patients with SWS is asymmetric, with decreased voltages and focal discharges in the affected brain hemisphere. EEG is also very useful for distinguishing migraines and seizure after cerebrovascular events as causes of the acute paroxysmal events.1
EEG in patients with SWS appears to change over time, becoming increasingly abnormal, with more frequent epileptiform activity.39
Perfusion ImagesDuring the first years of life, the vascular malformation tends to be hyperperfused. The pattern changes to hypoperfusion at the end of the first year of life, even in patients without seizures,24,37 although these changes are accelerated by crises and progressive ischemia, hypoxia, and glucose deprivation in the parenchyma underlying the malformation, and these are responsible for the progressive neurological deterioration in patients with SWS.
AngiographyAngiography is not routinely performed, but it can be useful in atypical cases to evaluate other associated vascular abnormalities (venous occlusion, thrombotic lesions, or arteriovenous malformations) or to identify large diploic vessels, thus avoiding bleeding in the event that craniotomy is necessary.24
DiagnosisDiagnosis of SWS is suspected in patients with PWS on the forehead (Fig. 8). Faced with this suspicion, ophthalmologic examination and contrast-enhanced MRI should be performed to enable early diagnosis and reduce ophthalmologic and cerebral complications.13 Imaging studies play an important role in diagnosis, detection, and follow-up of patients with SWS.
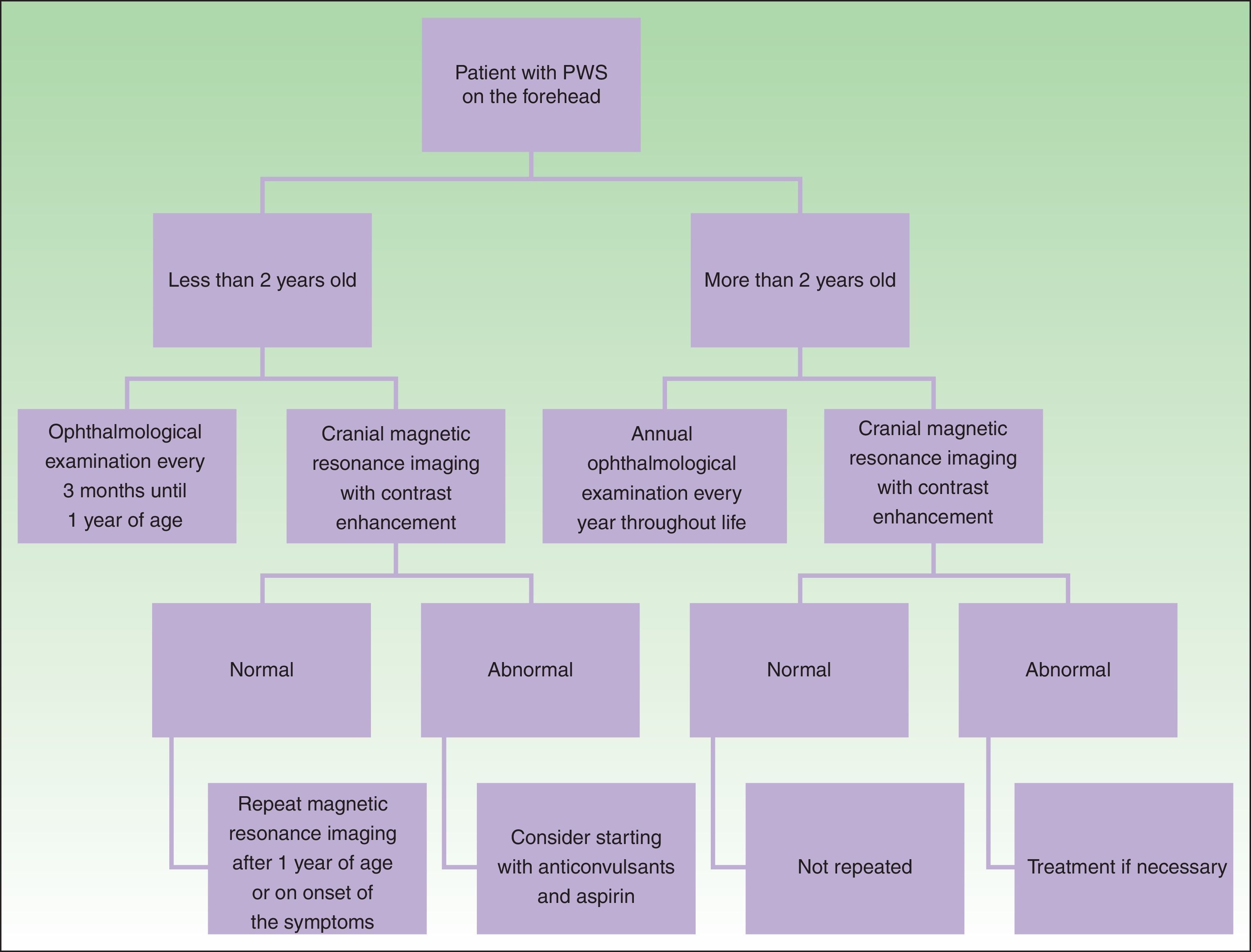
The need to perform cranial MRI in all patients with PWS at a site of risk is debated, given that there is a small percentage of false negatives and, on the other hand, not all patients with leptomeningeal involvement will present with seizures (10%).24 Given that the percentage of false negatives is small, and that detection of leptomeningeal angiomatosis may help orient parents for detection of seizures or actions to take when one occurs, we prefer to perform this study even in asymptomatic patients. Leptomeningeal angiomatosis may be hard to detect in the first 3 months of life, and so MRI is recommended when the patient is between 3 and 6 months of age.14
In the event that the patient presents with a PWS at a site of risk after 2 years of age, the need for MRI is more debatable in an asymptomatic patient because the patient is unlikely to have seizures that are difficult to control if no prior seizures have occurred.1
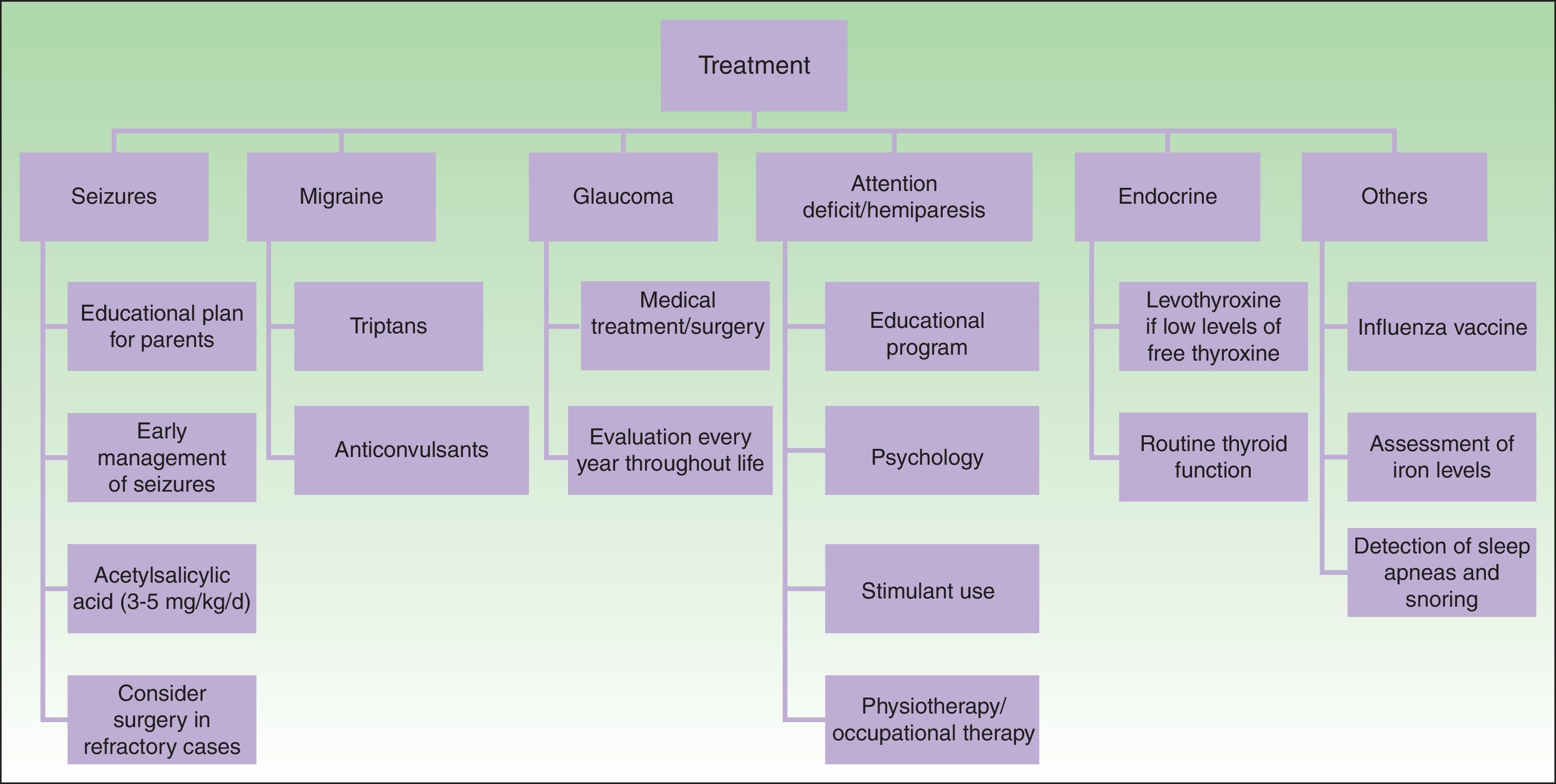
TreatmentMedical treatment in patients with SWS includes anticonvulsants, symptomatic and prophylactic treatment for headache, glaucoma treatment to reduce intraocular pressure, and laser therapy for PWS24 (Fig. 9).
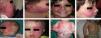
Treatment with pulsed dye laser (PDL) is the treatment of choice for facial PWS.40 In general, between 7 and 15 sessions are needed to clear the lesions, and rarely do the lesions completely disappear.41,42 The degree of response depends on the initial color of the lesion and where it is located. In general, PWS in the frontal region respond better than those in the malar region or prolabium43 (Fig. 10A, B, C, D, E, F, G, H). Most authors agree that responses are usually better if the treatment is performed during childhood rather than as adults, although there is no solid evidence to support this observation. In general, it is recommended to start treatment as soon as possible.43–46 Nevertheless, with a certain type of PWS, it is difficult to predict a priori the degree of response that will be achieved. Therapeutic resistance is multifactorial, but it is believed that this is partly due to regeneration and revascularization of photocoagulated vessels. Clinical trials have demonstrated that better responses are obtained with fewer sessions when rapamycin is administered, either topically or orally, immediately after treatment with PDL. Even so, the use of topical rapamycin for this indication is still not widespread in clinical practice, and the use of oral rapamycin does not appear to be supported for this indication.47,48
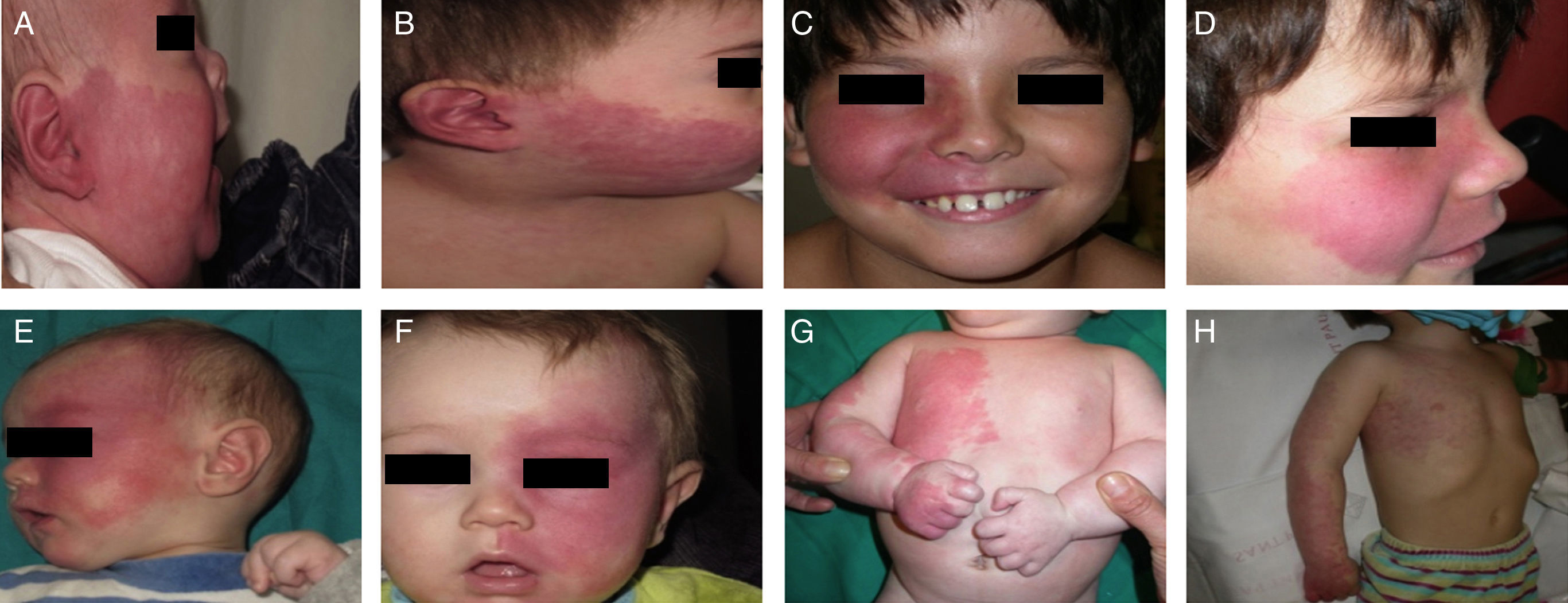
Before and after 7 sessions with laser therapy and difference in response according to site; A, B, C, D, Mandibular area and intense pink color in malar area, difficult to treat; E and F, frontal area and lateral area of face with better response than the central area; G, H, Area of limbs and trunk, worse response in acral areas. Note the good clearance in the pectoral area and arm.
Intense pulsed light, alexandrite laser light, sequential PDL light, and Nd-YAG laser light have also been used for treatment of PWS. Although these systems work well, treatment with PDL is still the treatment of choice.49
The key to preventing progression of neurological damage in SWS is early detection and management of seizures. A plan of action should be developed with the parents and local hospitals. It is important that the parents are instructed on how to recognize a seizure and how to act when faced with one. Moreover, given that fever or intercurrent diseases may trigger a seizure, it is important to control fever and ensure suitable hydration and control of oxygenation.1,23
Other important measures in the control of seizures in patients with SWS include: annual administration of influenza vaccine, increased dose of anticonvulsants according to weight, ruling out of iron-deficiency anemias, and detection and treatment of sleep apneas that may worsen cerebral ischema.14
Anticonvulsants remain the cornerstone of treatment of seizures. The anticonvulsants commonly used in nursing infants include carbamazepine, oxcarbazepine, levetiracetam, and phenobarbital.50 Carbamazepine and oxcarbazepine achieve better seizure control in patients with SWS.51
Given that seizures can lead to cognitive decline in patients with SWS, the usefulness of preventive treatment for seizures with antiepileptic agents has been considered, particularly in patients with extensive leptomeningeal involvement.50 The prophylactic use of phenobarbital before the first seizure is associated with a lower risk of cognitive decline compared with patients who receive the drug after their first crisis.52 This is a practice that cannot be generalized given the toxicity of phenobarbital, but it may be considered in patients with extensive bilateral cerebral involvement.50
Another potential prophylactic measure against neurological decline is acetylsalicylic acid at antiplatelet doses. Patients with SWS often have episodes of vascular ischemia resulting from venous stasis and thrombotic events. Some studies demonstrate that low doses of acetylsalicylic acid (3-5mg/kg/d) decrease the frequency and severity of cerebrovascular events and seizures.53,54 The adverse effects that can be observed with the use of acetylsalicylic acid are increased hematomas, nose bleeding, and gum bleeding.55
Seizures may be untreatable in 30% to 50% patients with SWS. In these cases, early surgery is indicated, either through lesionectomy or corpus callosotomy and hemispherectomy.1,23,50 In a study of 20 patients with SWS, it was demonstrated that hemispherectomy has an effectiveness of 90% in the elimination of seizures, and is more effective than focal resection.56 Some authors have reported the use of triptans and anticonvulsants for the treatment of headache.24
Patients with cognitive deficit and hyperactivity and attention disorders may benefit from specialist educational interventions, behavioral psychology, and stimulants (methylphenidate or dextroamphetamine).57 In patients with hemiparesis, it is important to initiate physiotherapy and occupational therapy as early as possible. Likewise, neuropsychological examination should be performed in older children.50,58
The main objective in glaucoma treatment in patients with SWS is to control intraocular pressure to prevent damage to the optic nerve.30 Treatment of early-onset glaucoma or glaucoma with camerular angle abnormalities is usually surgery, whether goniotomy or trabeculectomy. The success rate of angle surgery is generally lower than in primary congenital glaucoma, and further surgery, such as trabeculectomy or placement of a drainage device, is often required. The main complication of surgery is retinal hemorrhage when the decrease in intraocular pressure occurs too rapidly.50 Topical medication is used for the treatment of late-onset glaucoma. Aqueous suppressants and those that increase uveoscleral outflow tend to be the most effective.59,60 Newborn infants with a risk of glaucoma (frontal and periocular PWS) should be assessed by an ophthalmologist every 3 months for the first years of life; even if there is no evidence of glaucoma, annual eye examinations are recommended throughout the patient's life.14
Finally, it is important to monitor growth and symptoms of thyroid dysfunction in patients with SWS to detect growth hormone deficiencies and hypothyroidism.2,35
PrognosisPrognosis in patients with SWS depends mainly on the age of onset of the neurological symptoms. Early onset of seizures, lack of response to anticonvulsive therapy, extent of the leptomeningeal malformation, impact on perfusion of the brain cortex, and severity of ocular involvement all influence prognosis.
Ethical ResponsibilitiesProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of dataThe authors declare that they have followed their hospital's protocol on the publication of data concerning patients.
Right to privacy and informed consentThe authors declare that patient data do not appear in this article.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Higueros E, Roe E, Granell E, Baselga E. Síndrome de Sturge-Weber: revisión. Actas Dermosifiliogr. 2017;108:407–417.


