



Sr. Director:
El síndrome de Klippel-Trénaunay es una enfermedad de etiología desconocida muy poco frecuente que afecta a 1/27.500 recién nacidos1. Suele localizarse de forma unilateral en las extremidades, principalmente en las inferiores, y se caracteriza por una malformación vascular combinada o compleja (clasificación de la Sociedad Internacional para el Estudio de las Anomalías Vasculares) con trastornos venosos, linfáticos y capilares asociados a hipertrofia de tejidos blandos adyacentes y crecimiento óseo. El diagnóstico es fundamentalmente clínico, aunque en ocasiones son necesarios procedimientos de neuroimagen. Su curso es benigno pero progresivo y el tratamiento suele ser conservador, tratando las complicaciones derivadas de la enfermedad2,3. Se han descrito asociaciones con malformaciones vasculares y otras anomalías congénitas cardiovasculares, esqueléticas, digestivas o neurológicas. De forma excepcional se presenta junto con otros procesos cutáneos como la facomatosis pigmentovascular4.
La malformación de Arnold-Chiari es una enfermedad neurológica de causa desconocida que se cree debida a un desarrollo insuficiente de la fosa craneal posterior, con la consiguiente expansión del cerebelo en dirección al canal raquídeo. Se han descrito cuatro variantes de la malformación, siendo el tipo I que presentamos la más frecuente de todas, caracterizándose por el desplazamiento caudal del cerebelo con herniación amigdalina por debajo del foramen magno y alargamiento en forma de cuña de las amígdalas. La malformación de Arnold-Chiari es más frecuente en mujeres (1:3) durante la quinta década de la vida. Su diagnóstico se basa en los hallazgos característicos, anteriormente descritos, de la resonancia magnética. Los síntomas más comunes son la cefalea occipital, que se desencadena con la maniobra de Valsalva o con la extensión del cuello, dolor retroorbitario y trastornos visuales o cuadros simulando enfermedad de Ménière con hipoacusia, vértigo y tinnitus5. A su vez, la compresión del tronco encefálico puede dar lugar a hidrocefalia o siringomielia hasta en un 40 % de los casos6.
Presentamos el caso de una paciente de 54 años de edad con antecedentes de talasemia minor, intervenida de hernia discal L5-S1 y de un tumor óseo parietal derecho en 2000 del cual no aportaba informes. Fue diagnosticada de síndrome de Klippel-Trénaunay, tras valoración dermatológica, en 2003 ante la presencia de una malformación vascular tipo nevus flammeus en la extremidad inferior izquierda de límites bien definidos y geográficos, junto con venas varicosas en su interior que le conferían una tonalidad eritematoviolácea, todo ello acompañado de un alargamiento llamativo de dicha extremidad (figs. 1 y 2). Con posterioridad precisó valoración neurológica por un cuadro de inestabilidad y de giro de objetos de varios meses de evolución. En la exploración neurológica presentaba un signo de Romberg con caída a la derecha y marcha inestable a la deambulación. Se solicitaron pruebas de imagen donde se objetivó la presencia de un descenso de las amígdalas cerebelosas en el interior del foramen occipital compatible con malformación de Arnold-Chiari tipo I (fig. 3).

Figura 1.Malformaciónvascular en extremidadinferior.
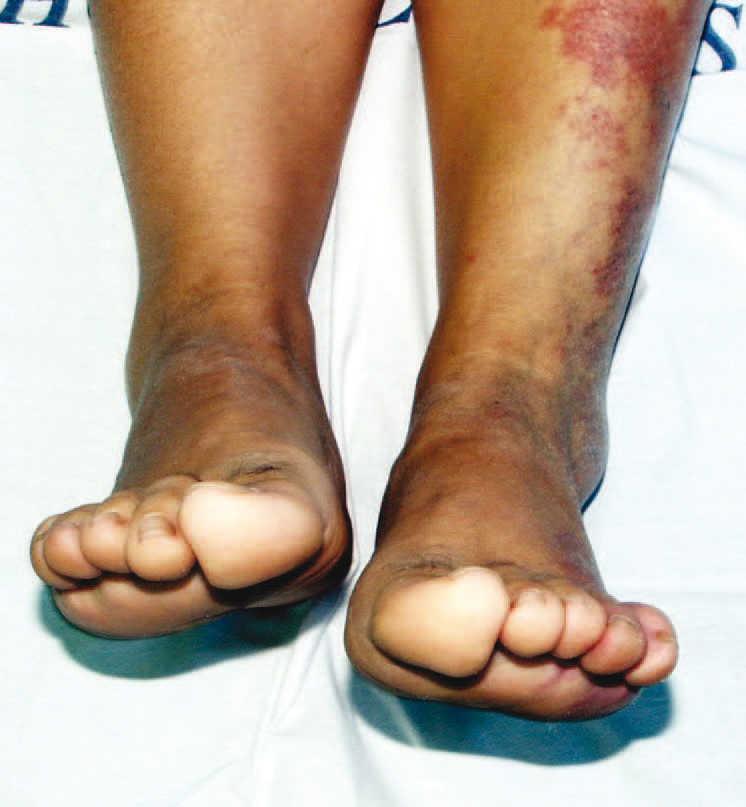
Figura 2.Alargamiento de la extremidadafectada.
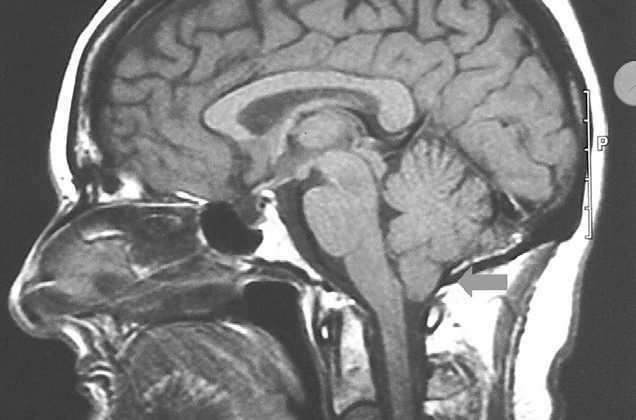
Figura 3.Herniación amigdalina por debajo del foramen magno.
En los trastornos neurológicos la asociación de síndrome de Klippel-Trénaunay y malformación de Arnold-Chiari es excepcional. Sólo hemos encontrado un caso publicado en la literatura revisada (MEDLINE 1966-2007)7. La baja frecuencia de ambos procesos descarta en cierta medida que dicha asociación sea de carácter casual, por lo que creemos importante que ante todo paciente con lesiones cutáneas de síndrome de Klippel-Trénaunay se investigue la presencia de clínica neurológica que haga sospechar una malformación de Arnold-Chiari, debiendo plantearse la realización de una resonancia magnética a todos estos pacientes.


