



INTRODUCCION
El síndrome de Kindler (OMIM 173650) es una forma infrecuente de genodermatosis, de herencia autosómica recesiva, que se caracteriza por la formación de ampollas ante mínimos traumatismos, especialmente durante la infancia y fotosensibilidad. La tendencia en la vida adulta es hacia el desarrollo de una poiquilodermia generalizada y atrofia cutánea, además de afectación de las mucosas oral y genitourinaria, con ocasional degeneración maligna 1, mientras que la fotosensibilidad suele mejorar con la edad.
Describimos a continuación los hallazgos en un varón de 33 años diagnosticado de síndrome de Kindler, con antecedentes de consanguinidad y otros miembros de la familia afectados posiblemente por el mismo problema.
DESCRIPCION DEL CASO
Un varón de 33 años refería, desde pocos meses después del nacimiento, la formación de ampollas en diversas localizaciones, fundamentalmente en las manos y en los pies, ante roces o traumatismos mínimos. Se acompañaba de una llamativa fotosensibilidad, que con el paso del tiempo fue presentando mejoría progresiva.
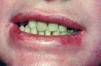
Las mucosas oral, digestiva y urinaria se vieron igualmente afectadas a lo largo de su infancia y juventud. En la boca, las alteraciones incluyeron destrucción de varias piezas dentarias, dificultando en su momento el tratamiento estomatológico y formación de rágades. En la mucosa digestiva, desarrolló una dificultad progresiva en la apertura de la boca junto con una disfagia de predominio a sólidos por formación de una membrana esofágica anterior, que precisó la realización de varias dilataciones a dicho nivel. Varios episodios de fimosis y balanitis originaron finalmente una estenosis en el meato urinario.
Entre los antecedentes familiares, destacaba el parentesco entre sus padres, que eran primos. Además, aunque no habían solicitado consulta dermatológica, 2 hermanas del paciente presentaron hallazgos clínicos semejantes. Una de ellas falleció a los pocos meses de vida por una sepsis, lo cual complicó un cuadro ampolloso.
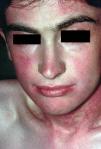
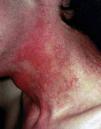
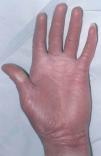
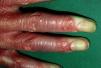
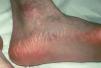
En el momento actual, a la exploración física el paciente presentaba placas poiquilodérmicas en la cara (figs. 1 y 2) y en los laterales del cuello (fig. 3), con áreas atróficas, telangiectásicas y esclerodermiformes. En ambas manos y pies, era evidente la esclerodactilia con afilamiento distal y cianosis digital, así como una leve onicodistrofia en las láminas ungueales (fig. 4). En la cara palmar era más llamativa la atrofia cutánea, con piel fina y lisa como papel de fumar y formación de pseudosindactilia (fig. 5). En las plantas de los pies, además, se apreciaban pápulas queratósicas o pits (fig. 6).

Fig. 1.--Atrofia, eritema y telangiectasias en mejillas y región cervical anterior.
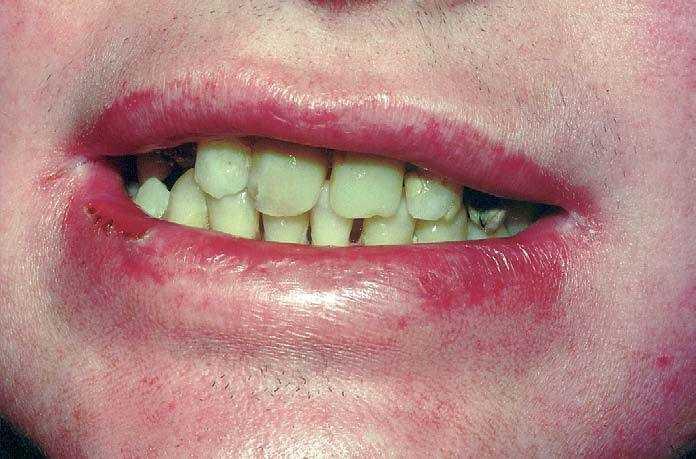
Fig. 2.--Deterioro moderado en piezas dentarias y rágades.

Fig. 3.--Placas poiquilodérmicas en laterales de cuello.
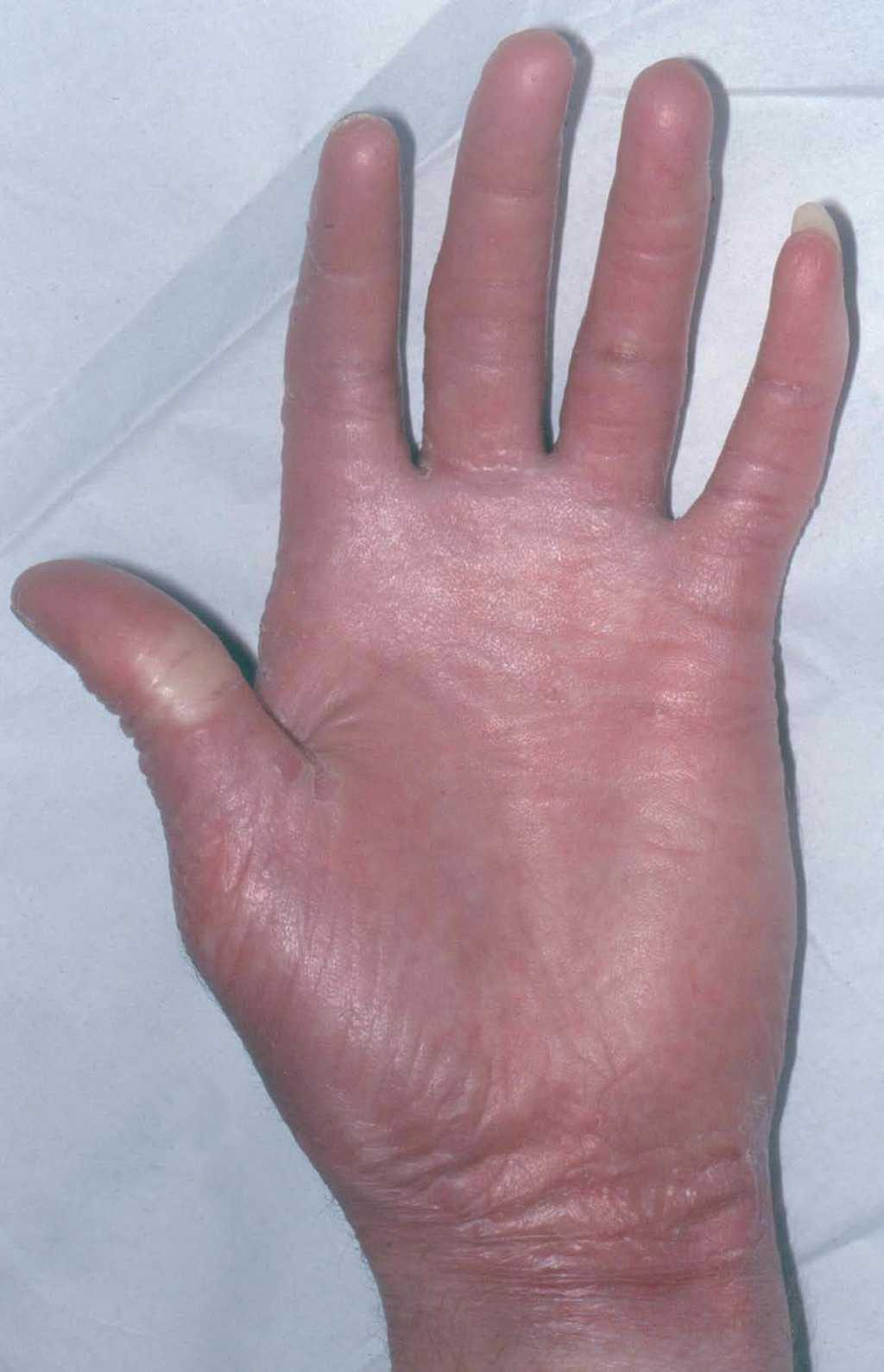
Fig. 4.--Atrofia cutánea y pseudosindactilia.
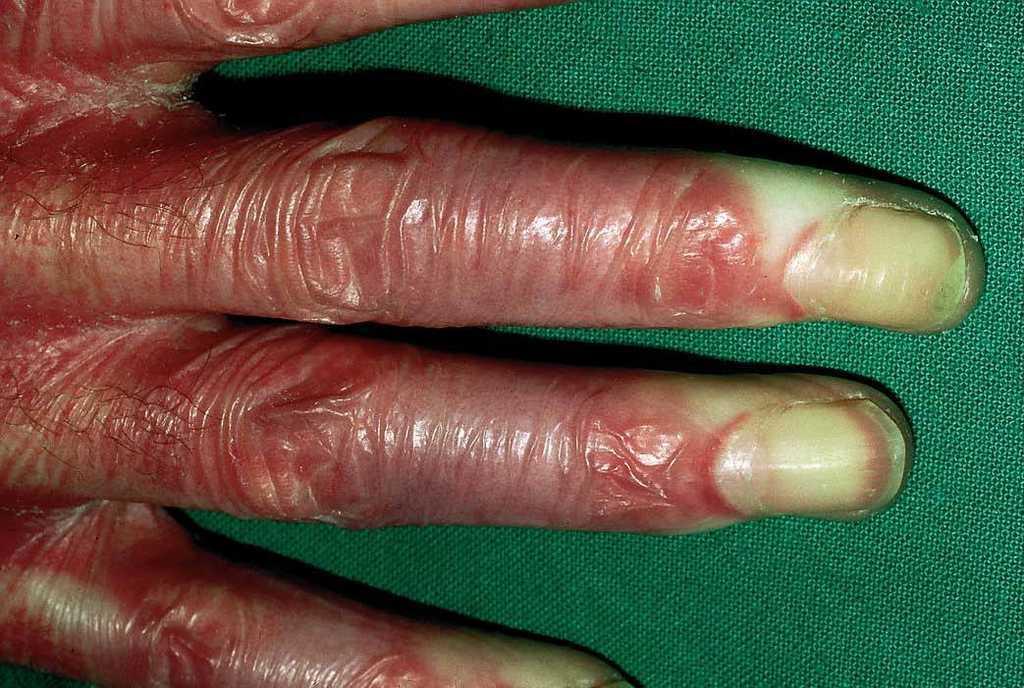
Fig. 5.--Esclerodactilia. Cianosis digital.
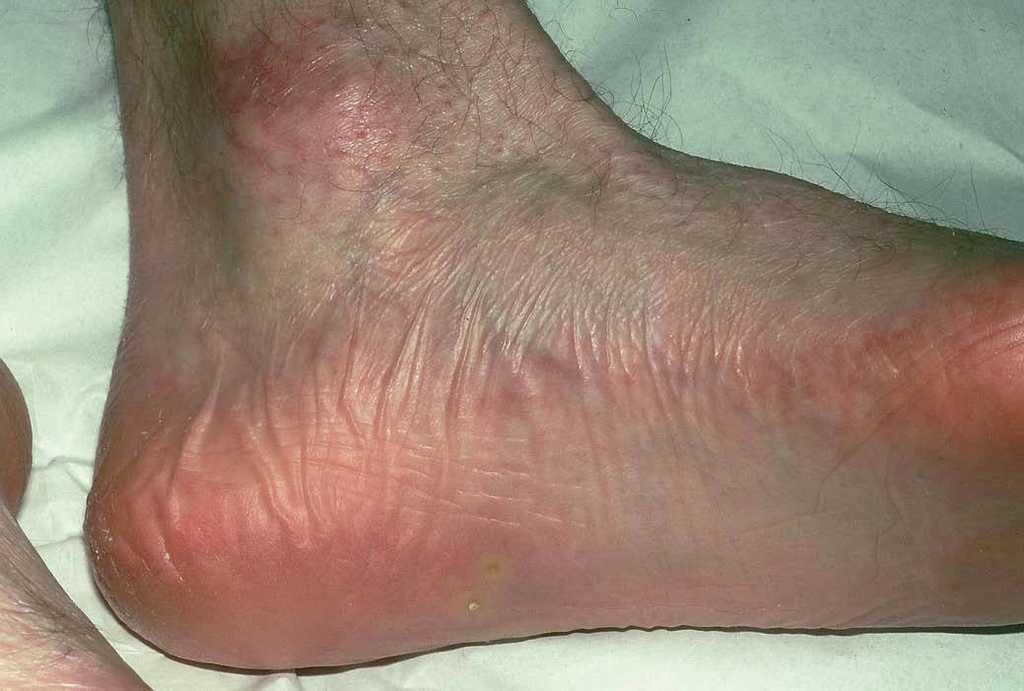
Fig. 6.--Hoyuelos plantares.
El estudio histológico de una de las lesiones demostró la existencia de intensa atrofia epidérmica, focos de degeneración hidrópica de la capa basal, dilataciones vasculares en dermis superficial y un mínimo infiltrado inflamatorio perivascular, cambios compatibles con los de una poiquilodermia.
El paciente ha realizado revisiones periódicas y se ha controlado de forma sintomática con medidas emolientes y fotoprotectoras. Se inició así mismo tratamiento con acitretina, en dosis de 25 mg/día, que se aumentó a 35 mg/día, con retirada de la misma 2 meses más tarde por escasa respuesta al tratamiento. Hasta el momento no se ha evidenciado desarrollo de degeneración maligna a ningún nivel.
DISCUSION
El síndrome de Kindler se considera una genodermatosis con entidad propia a pesar de combinar hallazgos clínicos de la epidermolisis ampollosa distrófica y de otras formas de poiquilodermia congénita 1,2. El patrón de herencia no está del todo claro. Aunque se han descrito casos esporádicos y otros de herencia autosómica dominante, la existencia de consanguinidad familiar apunta hacia una forma autosómica recesiva 3.
Recientemente se ha identificado el gen responsable del síndrome de Kindler a nivel del brazo corto del cromosoma 20 (20p12.3), y se han descrito mutaciones en un nuevo gen, KIND1, que codifica una proteína formada por 677 aminoácidos, denominada kindlin-1 (kindlerina). La kindlerina es un componente del citoesqueleto presente en los queratinocitos basales y que interviene en la unión, mediante contactos focales, en la actina con la matriz extracelular. Así, la mutación con pérdida en la función de este gen origina despegamientos entre los queratinocitos, fenómenos de reduplicación de la lámina densa, varios niveles de clivaje en la unión dermoepidérmica y alteraciones en las fibras elásticas 4-6. Todas estas alteraciones serían las responsables de las manifestaciones clínicas del síndrome, pero aún se desconoce el mecanismo exacto por el que las mutaciones originan la fotosensibilidad y la poiquilodermia 5. Se considera el síndrome de Kindler como la primera genodermatosis causada por un defecto en la unión actina-matriz extracelular a diferencia de la epidermolisis ampollosa, en la que se producen mutaciones en genes que codifican para filamentos intermedios de las queratinas 5 o 14 4,5.
Fue descrito por vez primera por Theresa Kindler en 1954 2, en una niña de 14 años que presentaba este tipo de alteraciones, que incluye la existencia de ampollas acrales neonatales o congénitas, y una marcada fotosensibilidad, con aumento en la susceptibilidad a las quemaduras, hallazgos que mejoran notablemente a lo largo de la infancia 3-10. La fragilidad cutánea y las ampollas son más llamativas en las zonas expuestas a traumatismos, sobre todo en las manos y en los pies, zonas en las que se pueden originar, como consecuencia, adherencias o sindactilias, acompañadas de distrofias ungueales e hiperqueratosis acrales 3-10. La curación de las ampollas no siempre origina cicatrices secundarias, aunque hay casos con cicatrización permanente o formación de quistes de milio transitorios. En la época prepuberal va apareciendo una hiperpigmentación reticulada progresiva, telangiectasias y atrofia en las áreas fotoexpuestas, con posterior extensión a zonas protegidas. En la vida adulta, el hallazgo predominante es la poiquilodermia, que ya persistirá con posterioridad 1,7-10. En la mayoría de los casos, durante la etapa prepuberal se van desarrollando los cambios poiquilodérmicos, si bien existen pacientes que ya desde el nacimiento o la primera infancia parecen presentar estas alteraciones 11. Las manifestaciones clínicas incluyen afectación de mucosas con estenosis esofágica, anal, uretral o desarrollo de ectropión, así como fragilidad gingival, con erosiones y alteraciones en la dentición 1,4-10. Otras asociaciones descritas incluyen hiperqueratosis u hoyuelos palmoplantares, onicodistrofia y fusión interdigital en manos y pies 3.
La mayoría de los cambios histopatológicos son inespecíficos o compatibles con poiquilodermia e incluyen atrofia epidérmica, vacuolización de la capa basal, ectasia capilar, incontinencia pigmentaria y edema en la dermis superficial 1,3,4,7-10. En estos pacientes es posible la degeneración maligna, con desarrollo de carcinoma espinocelular en labio inferior o paladar duro, e incluso carcinoma de células transicionales de la vejiga, por lo que se recomienda seguimiento clínico y examen físico de los sistemas respiratorio, digestivo y urinario 7.
El tratamiento es sintomático y preventivo, con curas antibióticas locales que eviten la sobreinfección de las lesiones ampollosas y prevengan la formación de cicatrices. Es fundamental evitar o protegerse adecuadamente del sol, para impedir la progresión de la poiquilodermia, en especial del componente atrófico. La fisioterapia y la rehabilitación pueden prevenir las contracturas y, en ocasiones, los pacientes pueden precisar apoyo psicológico 7.
El diagnóstico diferencial en la infancia temprana incluye la epidermolisis ampollosa (síndrome de Bart). La distribución acral de las ampollas acompañadas de atrofia cutánea serán datos más a favor del síndrome de Kindler. En niños a partir de los 5 años, en los que la poiquilodermia es más llamativa, se planteará el diagnóstico diferencial con los síndromes de Rothmund-Thomson y de Cockayne, fundamentalmente 3,7,8.
En 1971, Weary describió el síndrome que lleva su nombre, también denominado poiquilodermia hereditaria acroqueratótica 12. Se trata de un proceso casi superponible al del síndrome de Kindler en el que faltan la fotosensibilidad, la atrofia, las telangiectasias y las alteraciones mucosas. Típicamente, la cara está respetada, y es más evidente la despigmentación en las flexuras; se puede desarrollar una dermatitis eczematosa diseminada, ausente en el síndrome de Kindler 1,7,10. Algunos autores consideran que ambos síndromes constituyen variantes de una misma enfermedad, de hecho, se han descrito casos en los que se solapan características de ambos y a los que se les ha denominado síndrome de Weary-Kindler 1,10.
El diagnóstico diferencial de la poiquilodermia acroqueratótica y el síndrome de Kindler incluye todas las genodermatosis en las que se observa poiquilodermia como característica clínica prominente, incluyendo la poiquilodermia esclerosante hereditaria, el síndrome de Rothmund-Thomson, el xeroderma pigmentoso, la disqueratosis congénita, el síndrome de Mendes da Costa, la dermatopatía pigmentosa reticular, el síndrome de Franceschetti-Jadassohn y el síndrome de Degos-Touraine, fundamentalmente (tabla 1) 7.



