


INTRODUCCION
El xeroderma pigmentoso es una enfermedad genética multisistémica caracterizada por aumento de la sensibilidad a la radiación ultravioleta y alteración en la capacidad para reparar el ADN dañado por dicha radiación. Como consecuencia, los pacientes desarrollan neoplasias cutáneas y oculares que, con frecuencia se presentan ya en edades tempranas de la vida 1-3. El mecanismo patogénico no está claro, pero la alteración básica de esta enfermedad es la falta de capacidad para reparar de forma adecuada el ADN tras escisión del mismo debido a anormalidades en las endonucleasas 4. Recientemente se han relacionado también con la patogenia de la enfermedad alteraciones inmunológicas que implican a las células natural-killer5. Estudios de fusión celular, que consisten en la unión de poblaciones celulares distintas de 2 pacientes con xeroderma pigmentoso, demostraron la heterogeneidad de los defectos moleculares al conseguir corregir el defecto en la reparación del ADN que mostraban por separado 6. Desde entonces se han descrito 7 grupos de complementación diferentes (A-G) y una variante, caracterizados por poseer diferentes defectos enzimáticos, y que en cierto modo se corresponden con la gran diversidad clínica que puede presentar el xeroderma pigmentoso.
Además de las alteraciones cutáneas y oculares, también pueden asociar alteraciones neurológicas. La primera referencia a esta asociación fue aportada por Albert Neisser en 1883, que describió a 2 hermanos con xeroderma pigmentoso que asociaban graves defectos neurológicos. Posteriormente, De Sanctis y Cacchione publicaron un caso familiar de xeroderma pigmentoso asociado a degeneración neurológica progresivo, retraso en el crecimiento y desarrollo sexual inmaduro. Las alteraciones neurológicas características del síndrome de De Sanctis-Cacchione incluyen sordera neurosensorial, retraso intelectual, microcefalia, retraso en el desarrollo motor, hiporreflexia y/o arreflexia, ataxia y coreoatetosis. El síndrome de De Sanctis-Cacchione se ha relacionado con los grupos de complementación A y D, que son aquellos en los que la alteración del mecanismo de reparación es más grave 7, y se liga a una mutación en el gen ERCC68.
DESCRIPCION DEL CASO
Un niño de 4 años acudió a la consulta con su madre. Presentaba lesiones secundarias a quemadura solar localizadas en las mejillas, el dorso de la nariz y las orejas (fig. 1). La madre refería que, desde los primeros meses de vida, después de una breve exposición solar el paciente presentaba eritema y ampollas en las zonas expuestas. Cuando las quemaduras se resolvieron observamos en área facial numerosas máculas pigmentadas tipo efélides de morfología irregular (fig. 2). El número de efélides y su atipia morfológica fueron aumentando con el tiempo, como pudimos observar en sucesivas consultas. Los padres no referían historia familiar de fotosensibilidad ni otras enfermedades dermatológicas y tampoco existía cosanguinidad entre los progenitores.
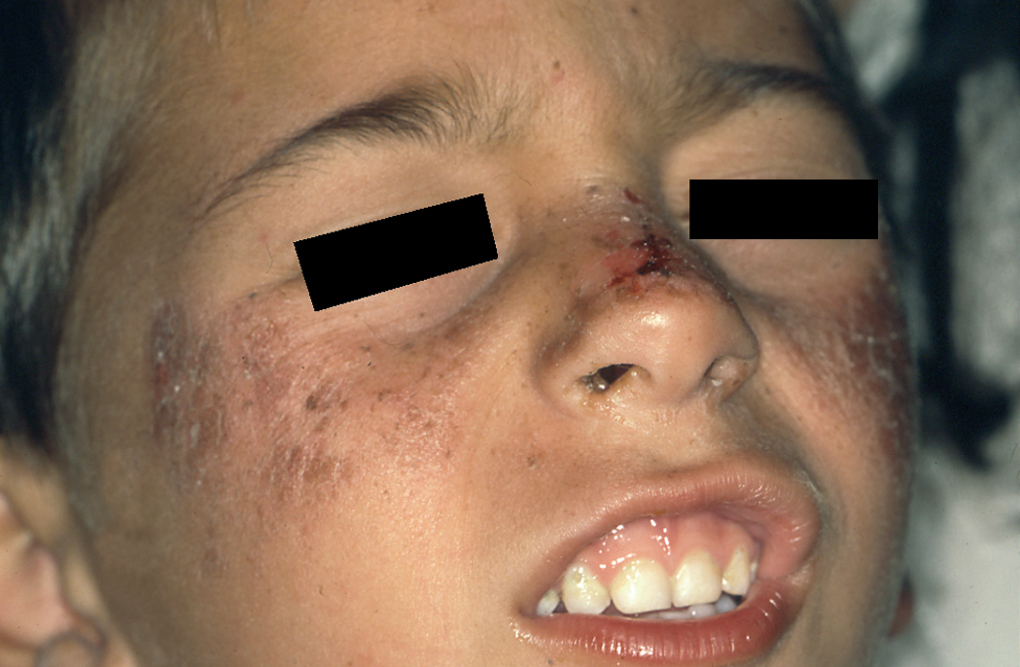
Fig. 1.--Lesiones faciales después de breve exposición solar.
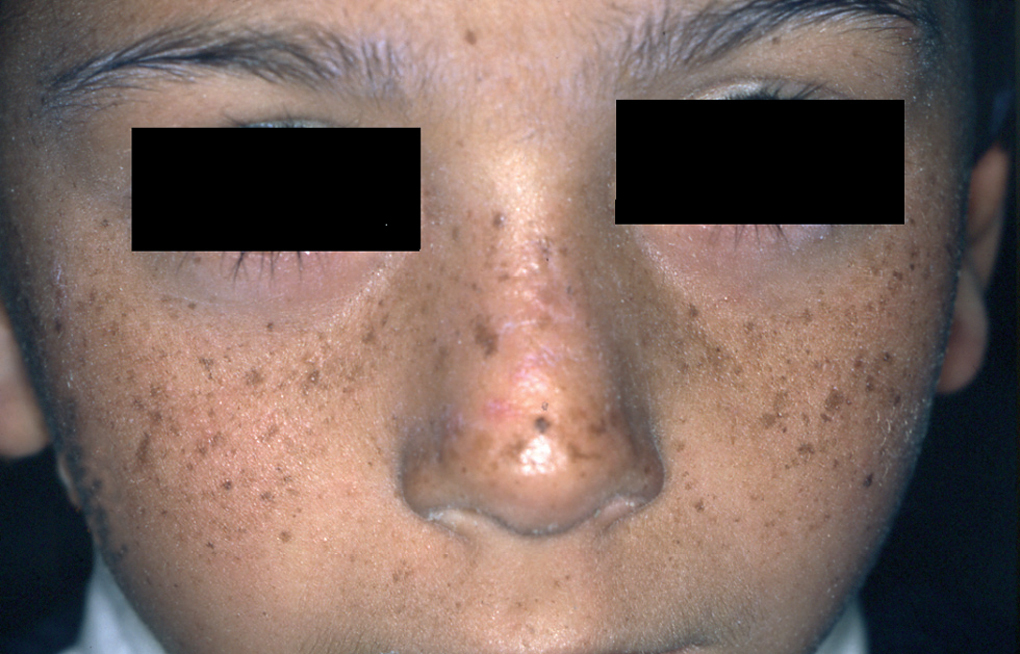
Fig. 2.--Numerosas lesiones tipo efélides de morfología irregular en zonas expuestas.
En el informe médico que se realizó tras el nacimiento constaba bajo peso y microcefalia, sin otros hallazgos relevantes. A los 2 años de edad ingresó en el servicio de Pediatría por retraso psicomotor y del crecimiento. Se realizaron estudios hormonales, serológicos y de imagen, que fueron todos normales a excepción de edad ósea retrasada radiológicamente.
En el momento de acudir a nuestra consulta observamos, en la exploración general, retraso ponderal, microcefalia, retraso en el desarrollo motor y mental, con notable retraso en el habla, arreflexia del tendón de Aquiles e hiporreflexia del resto de los reflejos. Se constataron también ocasionales movimientos coreoatetósicos y cierta alteración inespecífica de la marcha. Mediante estudio de potenciales evocados se detectó sordera neurosensorial con mínima expresividad clínica.
Los estudios de reparación de ADN demostraron niveles anormales de ADN reparado. Después de tratar los cultivos celulares con radiación ultravioleta C también se detectaron alteraciones en la síntesis de ARN (ambos estudios fueron realizados por el profesor A. R. Lehman, de la MRC Cell Mutation Unit, Universidad de Sussex, Reino Unido).
Se realizaron otros estudios de laboratorio, con el fin de descartar fundamentalmente otras causas de fotosensibilidad, incluyendo niveles de anticuerpos antinucleares, porfirinas, inmunoglobulinas y análisis cromosómico. No se detectaron alteraciones en ninguno de ellos a excepción de concentraciones de inmunoglobulina G por debajo de la normalidad.
DISCUSION
La presencia de fotosensibilidad en un paciente plantea inicialmente varios diagnósticos diferenciales; son esenciales estudios de laboratorio y pruebas especiales para un correcto diagnóstico. Sin embargo, algunas características clínicas pueden orientar con gran precisión el diagnóstico final. En nuestro caso, la presencia de fotosensibilidad, con quemaduras desde la infancia tras breve exposición solar, asociada a lesiones tipo efélides que aumentan en número según avanza la edad del paciente, hacen que la primera hipótesis diagnóstica, aun en ausencia de neoplasias cutáneas, sea la de xeroderma pigmentoso. Mención especial merece el diagnóstico diferencial con el síndrome de Cockayne y la tricotiodistrofia; ambas entidades también presentan alteración en la reparación del ADN, pero no existe tendencia al desarrollo de neoplasias. En el caso, como el que describimos, en que, por la edad del paciente aún no ha desarrollado patología tumoral, otras características clínicas ayudan al diagnóstico diferencial. En el síndrome de Cockayne es básico el retraso en el desarrollo y la disfunción neurológica, pero de manera característica los pacientes presentan patología ocular (retinopatía pigmentaria progresiva, cataratas, pupilas mióticas, etc.) y una apariencia física peculiar (ojos hundidos, implantación baja de las orejas y extremidades largas en flexión) 9. En la tricotiodistrofia la patología que presenta el cabello y otras displasias ectodérmicas marcan el diagnóstico 10.
Aunque el primer caso de xeroderma pigmentoso fue descrito en el libro de Hebra y Kaposi en 1874, el término de xeroderma pigmentosum fue acuñado posteriormente, en 1882, para enfatizar las alteraciones pigmentarias en una piel seca. Este dato clínico, asociado a la fotosensibilidad y al elevado riesgo de desarrollar neoplasias cutáneas y/u oculares es la característica clínica más relevante de la enfermedad.
En 1883 Neisser hizo referencia a la asociación del xeroderma pigmentoso con alteraciones neurológicas 3. En estudios recientes se observa que aproximadamente el 18 % de los pacientes presenta daños neurológicos asociados 1. Aunque las alteraciones neurológicas son independientes de la exposición solar, se relacionan también con el defecto de reparación del ADN. De hecho, se han encontrado también alteraciones en la reparación del ADN en otras enfermedades neurológicas como la esclerosis lateral amiotrófica y la enfermedad de Alzheimer. Además, Andrews et al 11 demostraron correlación entre el grado de sensibilidad de los fibroblastos a la radiación ultravioleta y la gravedad en las manifestaciones neurológicas, y consideraron que la reparación del ADN es necesaria para mantener la integridad funcional del sistema nervioso. Se ha visto que algunos pacientes asocian también retraso en el crecimiento con microcefalia, retraso ponderal e inmadurez sexual. Fueron De Sanctis y Cacchione quienes describieron por primera vez un paciente con estas características clínicas y con alteraciones del sistema nervioso que incluían sordera neurosensorial, retraso intelectual y en el desarrollo motor, hiporreflaxia y/o arreflexia, ataxia y coreoatetosis. Al ser un síndrome poco frecuente no existen grandes series en la literatura médica, por lo que los casos aislados que se publican son de especial interés. En nuestro país se han comunicado varios casos, algunos de ellos familiares 12-14. Estamos de acuerdo en la reflexión de Lambert et al 2 cuando hacen referencia a que sólo se deben incluir en el síndrome de De Sanctis-Cacchione a aquellos pacientes que presentan los datos clínicos referidos por los autores originales. Sin embargo, debido al curso progresivo de la enfermedad, es posible que no todos los datos clínicos del síndrome estén presentes en un momento determinado, ya que incluso, en ciertas ocasiones, las manifestaciones neurológicas no aparecen hasta la adolescencia 14. En nuestro caso, dada la edad del paciente, no se puede evaluar el desarrollo sexual, y por otra parte, aunque se observa cierta alteración de la marcha, no puede definirse claramente como ataxia. A pesar de ello, creemos que, al presentar asociado al xeroderma pigmentoso, retraso en el crecimiento, retraso psicomotor, sordera neurosensorial, coreoatetosis e hiporreflexia y arreflexia podemos diagnosticar a nuestro paciente de síndrome de De Sanctis-Cacchione.
De manera breve, queremos señalar que el tratamiento hasta la fecha es bastante desalentador. Se centra en la prevención de los tumores mediante fotoprotección cutánea y ocular, así como en el uso de retinoides orales (con la limitación de los efectos secundarios de esta medicación) y el abordaje terapéutico quirúrgico o médico de las lesiones tumorales ya desarrolladas. Destacan, en este aspecto, los buenos resultados que parecen obtenerse con imiquimod tópico al 5 % 15, así como con la asociación de este fármaco con acitretina oral 16. Nuevas terapias todavía experimentales, como el caso de la T4 endonucleasa V 17 abren nuevas perspectivas sobre un mejor futuro para estos pacientes.


