




Neurofibromatosis type 1 (NF1) is a common neurocutaneous syndrome that predisposes patients to the growth of several benign and malignant tumors, including malignant peripheral nerve sheath tumors (MPNSTs) and other soft tissue sarcomas.1 Liposarcoma in association with NF1 is very rare and just 10 cases have been reported in the literature.2
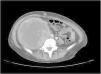
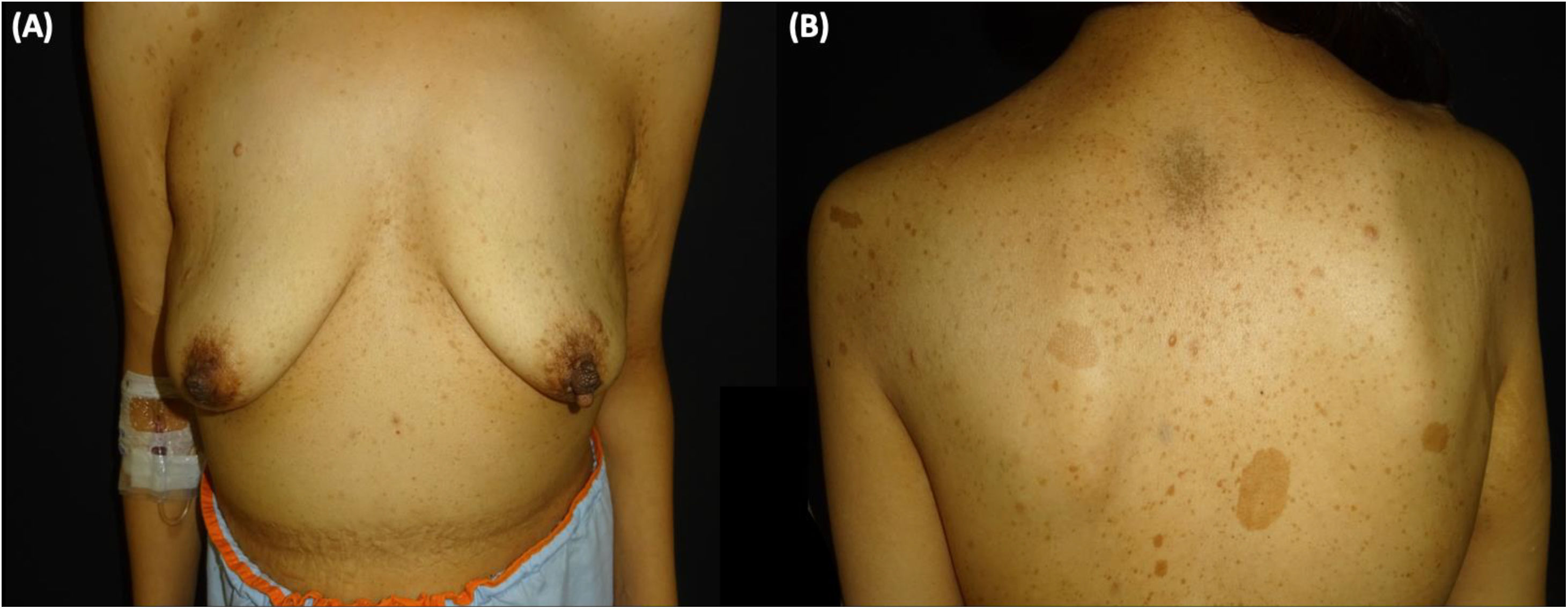
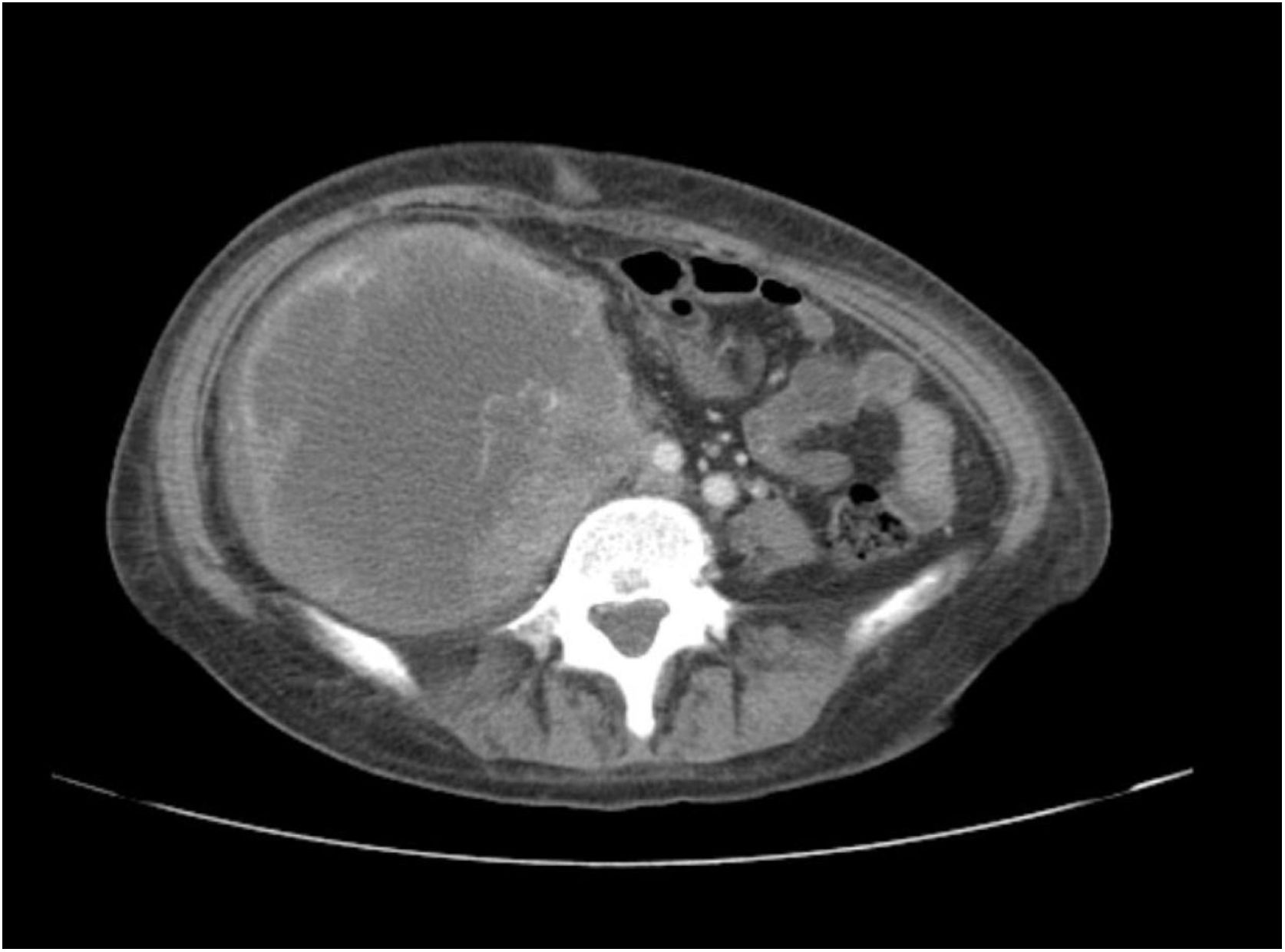
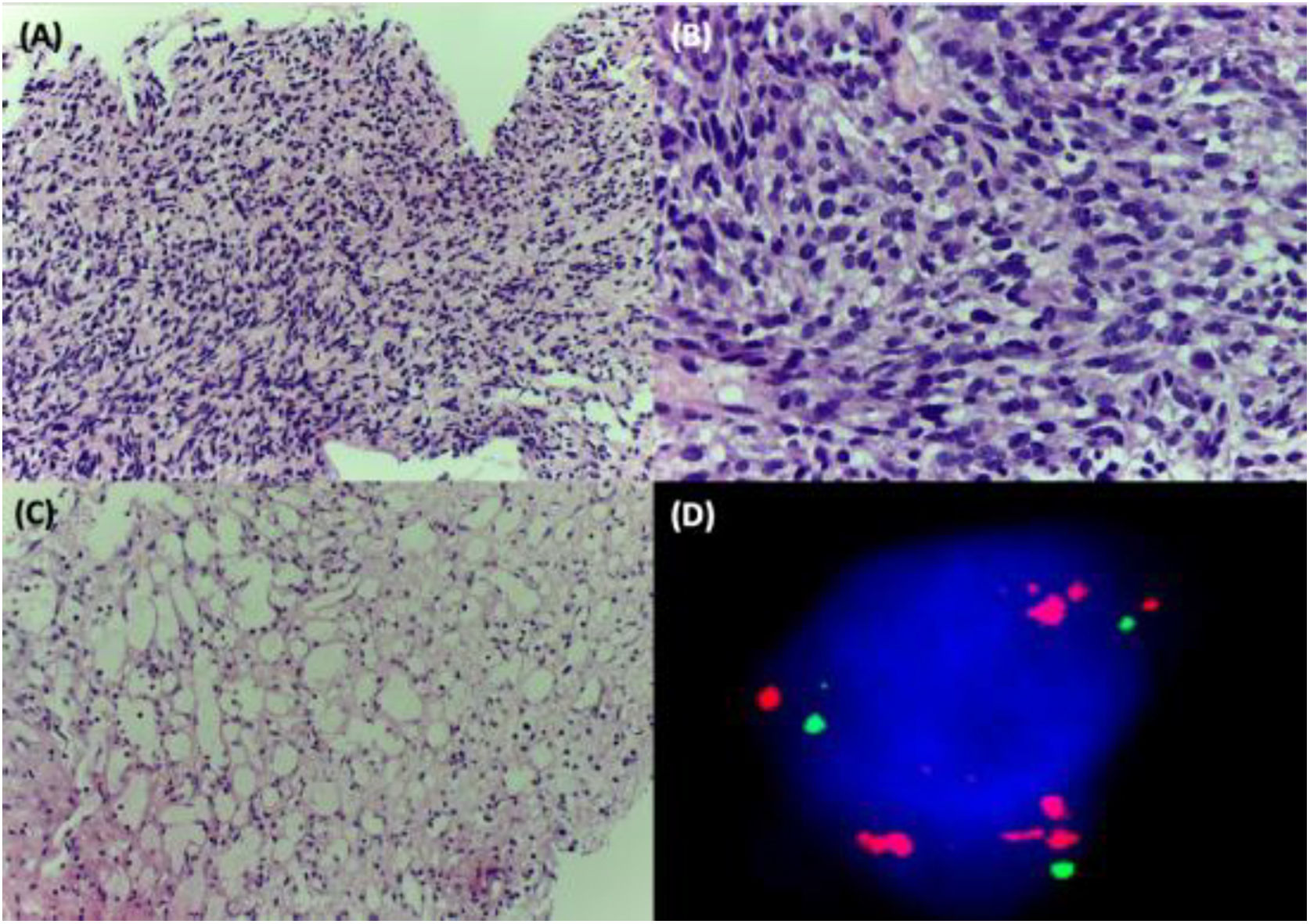
A 38-year-old woman with NF1 presented at the hospital’s emergency department with lower back pain of 3 months’ duration and constitutional symptoms. Physical examination of the abdomen revealed a large, hard mass on the right flank that was painful to the touch. She also had multiple café-au-lait spots, generalized freckles, mainly affecting the trunk and with a tendency to cluster in the area of the armpits, and several neurofibromas (Fig. 1). Computed tomography showed a right retroperitoneal mass with a diameter of 11 cm and multiple lung and brain lesions suggestive of metastasis (Fig. 2). Percutaneous biopsy of the mass showed a proliferation of spindle cells, extensive necrosis and abundant mitoses (Fig. 3A and B), and focal areas of increased cytoplasmic vacuolization consistent with adipocyte differentiation (Fig. 3C). Immunohistochemistry was positive for vimentin and CD99 only and negative for neural markers (S100, GFAP) and muscle markers (desmin and smooth muscle actin). Fluorescence in situ hybridization revealed MDM2 amplification in the long arm of chromosome 12 (Fig. 3D), leading to a definitive diagnosis of high-grade dedifferentiated liposarcoma.
Considering the extent of the disease, it was decided to treat the patient with palliative chemotherapy (adriamycin and olaratumab) and whole-brain radiotherapy. She was readmitted several months later because of clinical deterioration and poor pain control, and died shortly afterwards due to disease progression.
NF1 is caused by an alteration in the neurofibromin gene on the long arm of chromosome 17. The protein exerts a negative regulatory effect on the RAS/MAPK pathway, and loss of function activates the pathway, favoring the formation of tumors, which are the most serious complication of NF1. MPNST is the most common tumor in this setting. It occurs in 8% to 12% of patients and generally arises in an existing plexiform neurofibroma. Patients with MPNST are also more likely to develop other malignancies, such as pilocytic astrocytomas, gastrointestinal stromal tumors, pheochromocytomas, rhabdomyosarcomas, and juvenile myelomonocytic leukemia.1 Liposarcoma, however, is rare.2
Liposarcomas are malignant tumors with adipocyte differentiation; they are among the most common soft tissue sarcomas. There are 4 histologic subtypes—well differentiated, dedifferentiated, myxoid, and pleomorphic—each with considerable epidemiologic, cytogenetic, molecular, and prognostic differences. Dedifferentiated liposarcomas account for just 15% of all liposarcomas and tend to occur in the retroperitoneal space.3
Of the 10 cases of liposarcoma described in NF1 to date, 4 were pleomorphic, 2 were dedifferentiated, 2 were myxoid, and 1 was well differentiated. Histologic subtype was not specified in 1 case.2 This distribution of subtypes contrasts with that seen in the general population, where the most common subtype is well-differentiated liposarcoma (40%–50% of cases) and the least common, pleomorphic liposarcoma (< 10% of cases).3 The location of the tumors on the peritoneum or extremities, however, is similar to that observed in the general population.4,5 There have also been reports of liposarcomas involving the omentum,6 the temple,7 and the small bowel mesentery.2 Considering the few cases of liposarcoma described in patients with NF1, it is difficult to determine whether they differ from other liposarcomas in terms of epidemiology or prognosis. They may, however, be associated with a worse prognosis as the more aggressive histologic subtypes appear to be more common in this setting.
It should be noted that 4 of the liposarcomas described in association with NF1 in the literature arose in or adjacent to a pre-existing neurofibroma, giving rise to possible confusion with MPNST with liposarcomatous tissue. An estimated 15% of MPNSTs have heterologous elements, above all cartilage and bone, although they may also contain rhabdomyoblastic, squamous, glandular, and liposarcomatous tissue. Nonetheless, the pleomorphism observed in these 4 cases could be due to the multipotentiality of neurilemmal cells and their ability to undergo metaplasia to fat.7 Loss of the NF-1 tumor suppressor gene has been described in pleomorphic liposarcomas,3 and could explain the higher incidence of this more aggressive subtype in patients with NF1.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Bou Boluda L, Sabater Abad J, Quecedo Estébanez E, Jiménez Sánchez. Retroperitoneal Liposarcoma in a Woman With Neurofibromatosis Type 1. Actas Dermosifiliogr. 2021;112:856–858.


