



A wide range of treatments is now available for nonmelanoma skin cancer (NMSC), including 5-fluorouracil, ingenol mebutate, imiquimod, diclofenac, photodynamic therapy, methotrexate, cetuximab, vismodegib, and radiotherapy. All are associated with high clinical and histologic response rates. However, some tumors do not respond due to resistance, which may be primary or acquired. Study of the resistance processes is a broad area of research that aims to increase our understanding of the nature of each tumor and the biologic features that make it resistant, as well as to facilitate the design of new therapies directed against these tumors. In this article we review resistance to the authorized topical treatments for NMSC.
En la actualidad existe una amplia variedad de tratamientos para el cáncer cutáneo no melanoma (CCNM), como son 5-fluorouracilo, mebutato de ingenol, imiquimod, diclofenaco, terapia fotodinámica (TFD), metotrexato, cetuximab, vismodegib, radioterapia, todos ellos con altas tasas de respuesta clínica e histológica. Sin embargo, algunos tumores no responden al tratamiento, debido a la aparición de resistencias, tanto primarias como adquiridas. El estudio de los procesos de resistencia es un campo extenso de investigación que conlleva ampliar los conocimientos de la naturaleza de cada tumor, las características biológicas que lo hacen resistente y el diseño de nuevas terapias dirigidas contra los mismos. En el presente artículo se revisan las resistencias a los tratamientos tópicos autorizados para el CCNM.

There are many options available for the treatment of different types of nonmelanoma skin cancer (NMSC), including actinic keratosis (AK). This is important as surgery has its limitations and is not always feasible, such as when patients have multiple and/or extensive lesions or lesions in cosmetically sensitive areas. The introduction of chemotherapy drugs in recent years has increased the treatment options available and produced high complete response rates. Nonsurgical procedures have several advantages. In particular, they are noninvasive, offer excellent cosmetic results, and can be combined with other treatments and repeated. Examples of nonsurgical options used to treat NMSC are retinoids, 5 fluorouracil (5-FU), diclofenac, imiquimod, and photodynamic therapy (Table 1).1–13
Indications for Different Treatments Available for Nonmelanoma Skin Cancer, Including Approved and Off-Label Uses.
| Imiquimod 5%1,2 | 5-Fluorouracil1,3,4 | Topical 3% diclofenac gel and 2.5% hyaluronic acid1,5 | Ingenol Mebutate1,6,7 | Photodynamic Therapy1,8 | Vismodegib1,9 | Cetuximab10,11 | Intralesional Chemotherapy12,13 | |
|---|---|---|---|---|---|---|---|---|
| Approved uses (SPC) | Clinically typical nonhyperkeratotic, nonhypertrophic AKs on the face or scalp in immunocompetent adult patients | Slightly palpable and/or moderately thick hyperkeratotic AK (grade I/II) in immunocompetent adults (at concentration of 5mg of fluorouracil and 100mg of salicylic acid) Multiple AKs (at concentration of 5%) | AK | Nonhyperkeratotic and nonhypertrophic AKs in adults | AK | |||
| sBCC | sBCC | sBCC Nodular BCC | Symptomatic metastatic BCC Locally advanced BCC inappropriate for surgery or radiotherapy | |||||
| Bowen disease | ||||||||
| Other conditions | AKs in other locations and in immunodepressed patients Nodular BCC Bowen disease Keratoacanthoma Paget disease Erythroplasia of Queyrat SCC | Bowen disease | Bowen disease | BCC Bowen disease | SCC Keratoacanthoma Erythroplasia of Queyrat Paget disease Gorlin syndrome | Gorlin syndrome | Locally advanced, unresectable, or metastatic SCC Unresectable BCC in monotherapy or associated with smoothened inhibitors to reduce resistance | Methotrexate: keratoacanthoma, SCC Bleomycin: AK, sBCC, and Bowen disease Interferon alfa-2, alfa-2a, and alfa-2b: BCC, SCC, and AK Interferon β and γ: BCC |
Abbreviations: AK, actinic keratosis; BCC, basal cell carcinoma; sBCC, superficial basal cell carcinoma; SCC, squamous cell carcinoma.
The effectiveness of the above treatments, however, is limited by treatment resistance. Tumor cell resistance is defined as an absence of sensitivity to anticancer drugs and it has multiple, complex causes. Resistance is the main reason why anticancer drugs fail and it has an important role in tumor progression and poor prognosis. Although resistance to chemotherapy and radiotherapy has been extensively studied, we are still far from understanding the mechanisms involved. Generally speaking, the first treatment a patient receives destroys the majority of tumor cells, but if the tumor does not respond adequately to this treatment, resistant cancer cells will remain and may even become more aggressive after several treatment cycles.14
Resistance can be generally classified as intrinsic or acquired. Intrinsic resistance is characterized by the presence of pre-existing factors that influence how the tumor cells will respond to treatment, while acquired resistance develops after the treatment of a priori sensitive tumors. Intrinsic resistance is a complex process related to diverse biochemical and molecular features of the tumor that allow certain cells to avoid death. Acquired resistance, by contrast, can be caused by different factors, including the limited amount of drug or radiation that reaches the tumor, factors in the tumor environment, and possible mutations that arise in tumor cells during treatment.15
Additional adaptive responses also need to be considered, such as increased expression of the therapeutic target and activation of alternative compensatory signaling pathways. Cross-resistance is another problem, as once treated, tumors can develop resistance to other drugs, as occurs in multidrug resistance. Finally, certain tumors are highly heterogeneous and contain cells with different phenotypic, genetic, and/or epigenetic characteristics, meaning that sensitivity to treatment will vary according to the area of the tumor.16–18
In this article we offer an overview of resistance to nonsurgical treatments in NMSC based on a review of case reports and series and research into the possible mechanisms involved. Studies of resistance will contribute to a better understanding of tumor biology and will help to determine how best to combine treatments to improve response rates and reduce adverse effects.19
In the first of 2 articles, we will review work on possible mechanisms of resistance to the following topical treatments for NMSC: 5-FU, imiquimod, diclofenac, and ingenol mebutate.
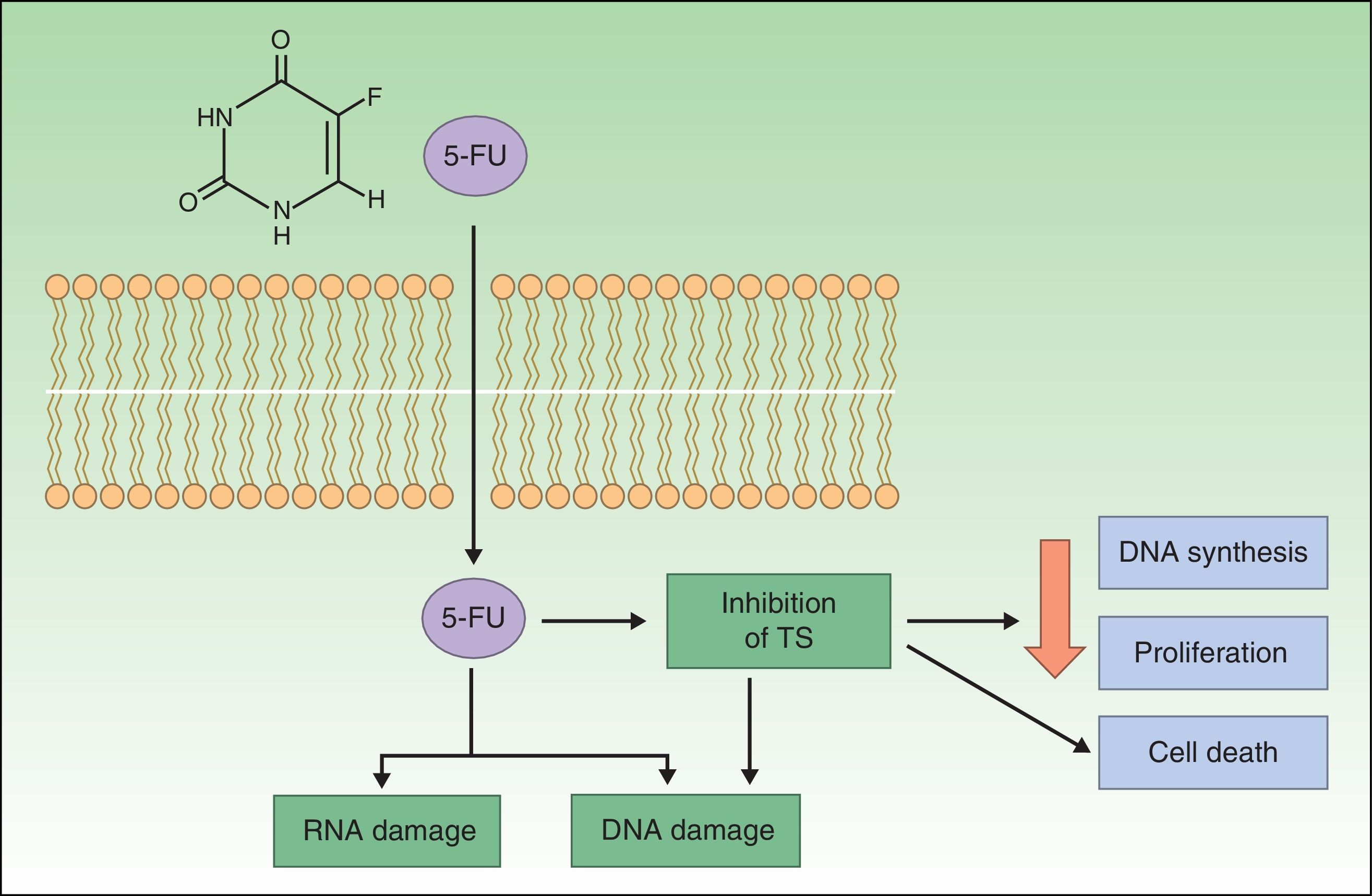
Resistance to 5-FU5-FU is a fluoropyrimidine that acts as an antimetabolite by binding to the enzyme thymidylate synthase, which is responsible for the synthesis of nucleotides. The resulting inhibition of thymidylate synthase leads to a reduction in DNA synthesis and cell proliferation, inducing cell death. These effects are particularly evident in cells with high mitotic rates, such as neoplastic cells. 5-FU is also incorporated into DNA or RNA, interfering with their normal functioning (Fig. 1).20–22
, which binds to and inhibits the enzyme thymidylate synthase (TS), thereby reducing DNA synthesis and cell proliferation and inducing cell death.")
Topical 5-FU is approved for the treatment of AK at concentrations of 0.5%, 1%, 2%, and 5%. The 5% formulation, applied twice daily for at least 6 weeks, is indicated for superficial basal cell carcinoma (BCC) and is associated with an approximate cure rate of 93%. Topical 5-FU is not indicated for the treatment of Bowen disease.22–24
In the largest series to date supporting the efficacy of topical 5-FU in the treatment of superficial BCC, a histologic cure rate of 90% was reported after 3 weeks for 31 superficial lesions treated twice daily for 11 weeks. Although the follow-up time was short, a treatment resistance rate of 10% was observed.23,25 Topical 5-FU is much less effective in nodular BCCs, and its use in this setting has had limited success.26
In the case of AK, 5% 5-FU cream applied for 2 to 4 weeks produced a clinical cure rate of 96% and a histological cure rate of 67%, although 54% of tumors had relapsed at 12 months.27
Although 5-FU is not approved for the treatment of squamous cell carcinoma (SCC), its efficacy in this setting has been analyzed in several studies. In one of these, 29 patients with SCC in situ were treated with 5-FU cream (Efudix) for 4 weeks. The cream was applied once a day for the first week and twice a day for the remaining weeks. Three months after the last treatment, a complete response rate of 83% was observed but at the 12-month follow-up, this had fallen to 69% and was accompanied by a recurrence rate of 17%.28 In another study of 26 patients with Bowen disease treated with 5% 5-FU cream twice a day for 9 weeks, 92% of patients achieved complete clearance over a mean follow-up period of 55 months.26
The above results clearly show that numerous NMSC lesions are resistant to treatment with 5-FU. However, in order to be able to predict—and resolve—resistance problems, it is essential to understand the mechanisms by which 5-FU induces apoptosis and why certain tumors do not respond.20
One study described the case of a patient with severe dihydropyrimidine dehydrogenase (DPD) deficiency who developed severe gastrointestinal and hematological toxicity following treatment with a standard dose of 5-FU for BCC. DPD is the first enzyme involved in the degradation of 5-FU.29 Approximately 10% of topical 5-FU is absorbed through the skin while over 80% is inactivated in the liver by DPD, explaining why its deficiency causes toxicity. In the case of colorectal cancer, however, patients with low DPD levels respond better to 5-FU, suggesting that DPD alterations and polymorphisms could be one cause of resistance.29
Increased expression of the Bag-1 protein has been observed in progressive, metastatic oral SCC. This protein has an antiapoptotic function associated with the 70-kDa heat shock protein (Hsp70), indicating that overexpression of these 2 proteins would increase tumor cell resistance to apoptosis.30 In one study, the elimination of Bag-1 from the cutaneous SCC cell line SCC-13 was found to increase sensitivity to 5-FU-induced apoptosis.31 The same study demonstrated overexpression of both Bag-1 and Hsp70 in a series of tumors, leading the authors to hypothesize that resistance to 5-FU in SCC might be mediated through a cytoplasmic Hsp70-dependent mechanism.
A final theory on 5-FU resistance is related to cancer stem cells in tumors of epithelial origin. According to this theory, malignant tumors, just like normal epidermis, would contain “stem cells” responsible for proliferation that would give rise to more differentiated tumor cells that would form the bulk of the tumor. Like regular stem cells, cancer stem cells have a slow cell cycle. It is therefore considered that they might be responsible for resistance to classic chemotherapy drugs that typically target proliferating cells.32
Key point: PDP alterations and polymorphisms and overexpression of Bag-1 and Hsp70 could influence sensitivity to 5-FU treatment.
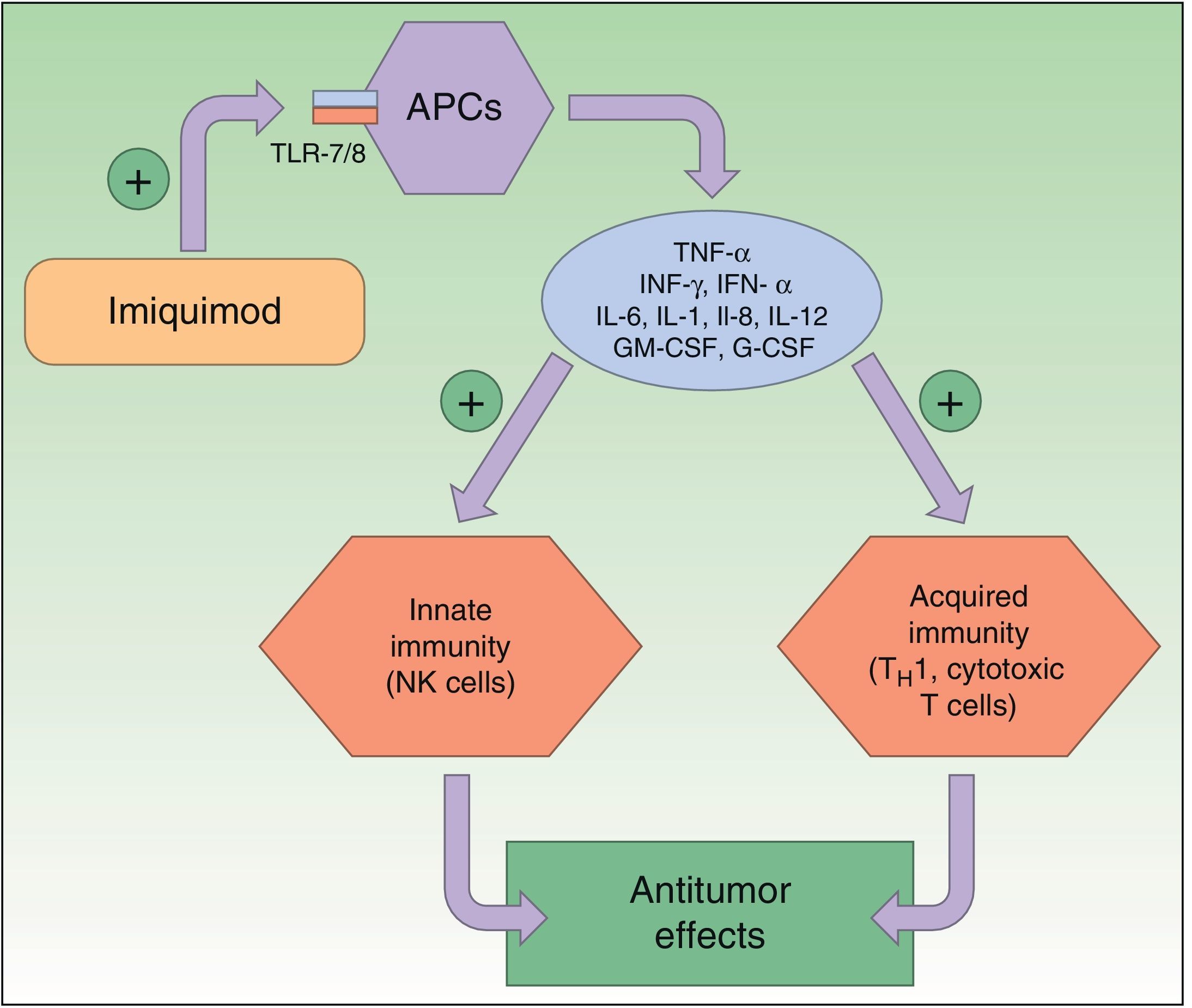
Resistance to ImiquimodImiquimod is a synthetic compound of the imidazoquinoline family that acts as an immunomodulator, stimulating both innate and acquired immune responses. The immune response is modified through the toll-like receptor 7 (TLR7) and TLR8 pathways; these receptors are located on the surface of antigen-presenting cells, such as dendritic cells, macrophages, Langerhans cells, etc. The activation of these pathways triggers the production and release of numerous cytokines and chemokines, tumor necrosis factor (TNF) α, interferon (IFN) γ, certain interleukins (ILs), and granulocyte-macrophage colony-stimulating factor, and attracts natural killer (NK) cells, thereby eliciting an innate and acquired immune response (Fig. 2). Imiquimod is thus a potent antiviral and antitumor agent, and is used widely in the field of dermatology, particularly in the treatment of malignant cutaneous lesions.2,26,33–37

Mechanism of action of imiquimod. This immunomodulator acts by blocking TLR7 and TLR8, triggering the release of proinflammatory and antimicrobial cytokines and stimulating innate and acquired immunity, with antitumor effects. APCs indicates antigen-presenting cells; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; INF, interferon; NK, natural killer; TH1, type 1 helper T cells; TLR, toll-like receptor; TNF, tumor necrosis factor.
Numerous studies have shown that imiquimod also inhibits the growth of new blood vessels thanks to its antiangiogenic properties. It induces an increase in IL-10 and IL-12 levels, which inhibit angiogenesis, reduce cell production of proangiogenic factors (such as fibroblast growth factor and IL-8), inhibit vascular motility, and induce endothelial apoptosis.26 There is also evidence that imiquimod induces keratinocyte apoptosis, thereby favoring cytochrome C release and caspase 3 activation.38
Imiquimod 5% cream is approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of external genital warts, superficial BCC, and AK.33,39 It is applied 3 to 5 times a week for 4 to 16 weeks depending on whether it is used to treat AK or BCC.40
Imiquimod has also been used to treat other types of NMSC, such as Bowen disease, Bowenoid papulosis, extramammary Paget disease, melanoma in situ, and keratoacanthoma, among others.38
Because surgery generally produces better results than topical treatments in skin lesions, imiquimod is mainly used in patients who are not candidates for surgery.33
Gupta et al.41 undertook a meta-analysis of 4 studies involving 393 patients to evaluate the effectiveness of imiquimod 5% for the treatment of AK, and described an average efficacy rate of 70% (with a 95% confidence interval of ±12%). A later study of 479 patients investigated the efficacy of the 2.5% and 3.75% formulations applied once a day for two 2-week periods separated by 2 weeks with no treatment. After 8 weeks of follow-up, the respective complete and partial response rates were 30.6% and 48.1% for imiquimod 2.5% and 35.6% and 59.4% for imiquimod 3.5%.42
Waalboer-Spuij et al.37 undertook a clinical trial in which 118 patients with AK were treated with imiquimod 5% once daily for 3 days a week over a month. After this period, 58% of patients required a second monthly cycle due to a lack of response. After 16 weeks of follow-up, the complete and partial response rates were 46% and 35%, respectively.
Very little is known about the efficacy of imiquimod in SCC, as it is not approved for this condition. In one study, curettage followed by application of imiquimod resulted in a 95% response rate at 12 weeks.43 In another study in which imiquimod only was applied for 9 to 12 weeks, the response rates were 71% for SCC and 57% to 80% for Bowen disease.40
Numerous studies have analyzed the use of imiquimod 5% in BCC using different application regimens, ranging from twice-daily to twice-weekly application, with follow-up times of 1 to 5 years. The cure rates oscillated between 42% and 100% and the most effective regimen was the twice-daily application. Most recurrences were seen in the first or second year after treatment.44,45
Cure rates for imiquimod are lower in the case of nodular BCC, with one study reporting a rate of 85.6% at 12 months for a regimen in which imiquimod was applied daily for 12 weeks.46
The TLR7 gene, located on chromosome X, has been investigated as a possible factor in resistance to imiquimod. In a study of 34 patients with BCC (28 responders and 6 nonresponders), Piaserico et al.47 reported that the presence of the T allele for the TLR7 polymorphism rs179008/Gln11Leu might be a resistance factor. Hemizygous males carrying this polymorphism have been found to have lower levels of TNF-α following imiquimod stimulation.48
Key point: Certain polymorphisms in the TLR7 gene might cause resistance to imiquimod, and reduced TNF-α levels have been proposed as a possible mechanism.
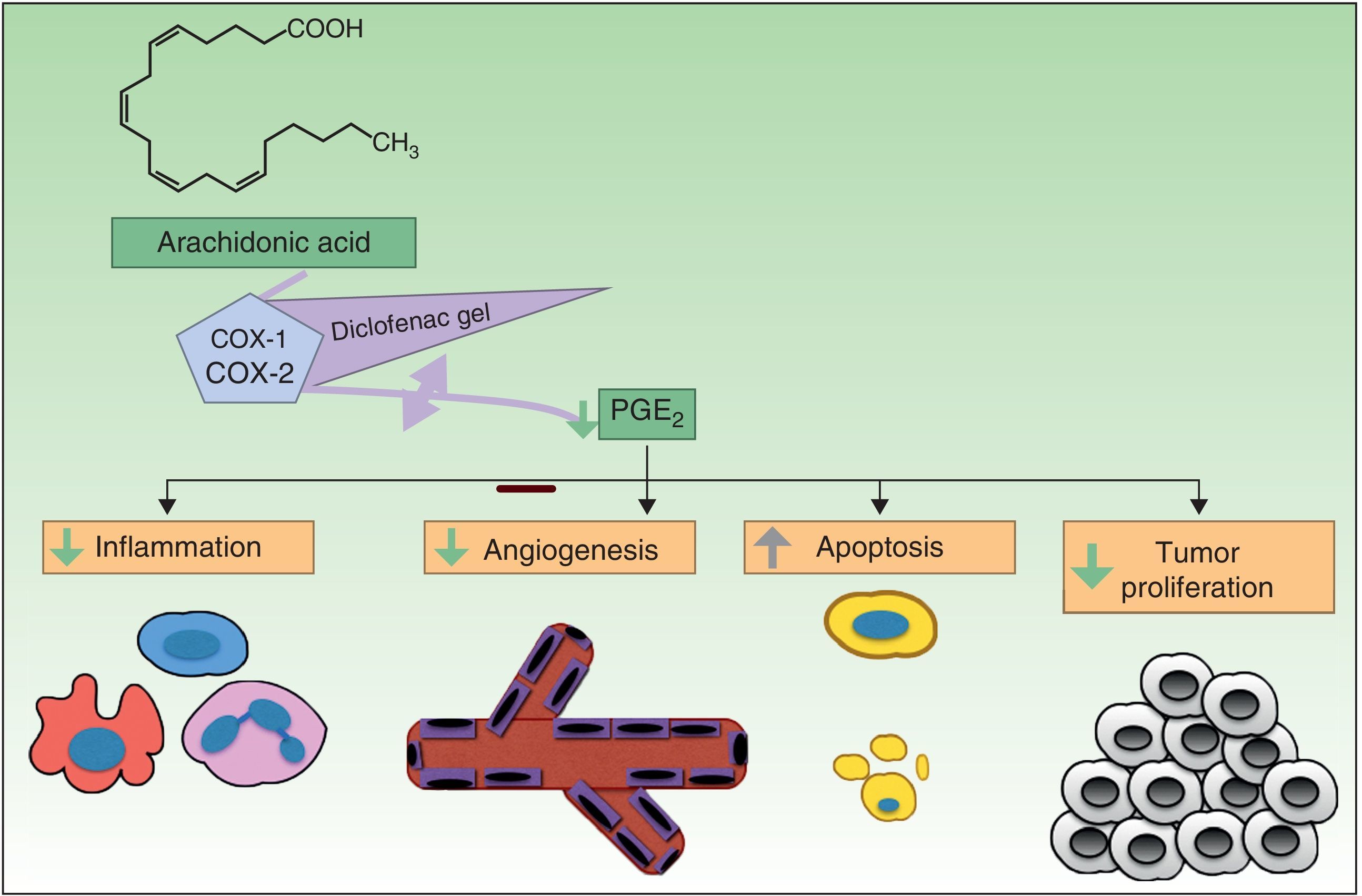
Resistance to DiclofenacDiclofenac is a nonsteroidal anti-inflammatory drug that reduces the production of prostaglandins through inhibition of cyclooxygenase 2 (COX-2) (Fig. 3). There is evidence that COX-2 has an important role in the development and progression of NMSC. COX-2 permits the formation of prostaglandin E2 (PGE2), which in turn enhances tumor proliferation, angiogenesis, and inflammation, and inhibits apoptosis. COX-2 inhibition is thought to achieve the opposite effect, but its mechanism of action in skin cancer cells is unknown. Diclofenac 3% in hyaluronic acid 2.5% (Solaraze) is available as a topical gel approved by the FDA and EMA for the treatment of AK. It is applied twice daily for 60 to 90 days. Although there are studies supporting the use of topical diclofenac in AK,49–52 and to a lesser extent Bowen disease,53,54 there are no data on its effectiveness in the treatment of BCC or invasive SCC.40
, favoring apoptosis. COX indicates cyclooxygenase; PGE2, prostaglandin E2.")
Complete response rates for AK treated with diclofenac vary widely, with figures ranging from 33% to 81% depending on the study, and there is even one study in which diclofenac failed to produce clinically significant improvements in 130 patients.55–59 There are therefore patients who do not respond to diclofenac and/or who develop recurrences.
The mechanisms of action underlying diclofenac resistance in AK are not clear. Considering the similarities between AK and SCC (mutated p53 and overexpression of COX-2), Rodust et al.60 used 4 cutaneous SCC cell lines as a model to study resistance to diclofenac in AK. Three of the lines were sensitive to the proapoptotic effects associated with diclofenac-induced caspase activation, while the fourth was resistant. Treatment of diclofenac-sensitive cells produced the characteristic proapoptotic effects at the level of the B-cell lymphoma proteins (Bcl-2) and resulted in the increased expression of Bad (proapoptotic) and the decreased expression of myeloid cell leukemia 1 (Mcl-1) and Bcl-w (both antiapoptotic). However, in the resistant line, the lack of COX-2 prior to treatment with diclofenac was already associated with low levels of Mcl-1 and Bcl-w and high levels of Bad, possibly due to the lack of PEG2 in the cells. In such a situation, diclofenac would be unable to exert its proapoptotic effects. However, these resistant cells were also seen to contain underexpressed levels of Noxa and Puma, 2 proapoptotic members of the Bcl-2 family, overall, possibly favoring a COX-2-independent antiapoptotic response to diclofenac.
Key point: The lack of response to diclofenac in SCC cells appears to be independent of pathways that modulate apoptosis through COX-2 in SCC cells.
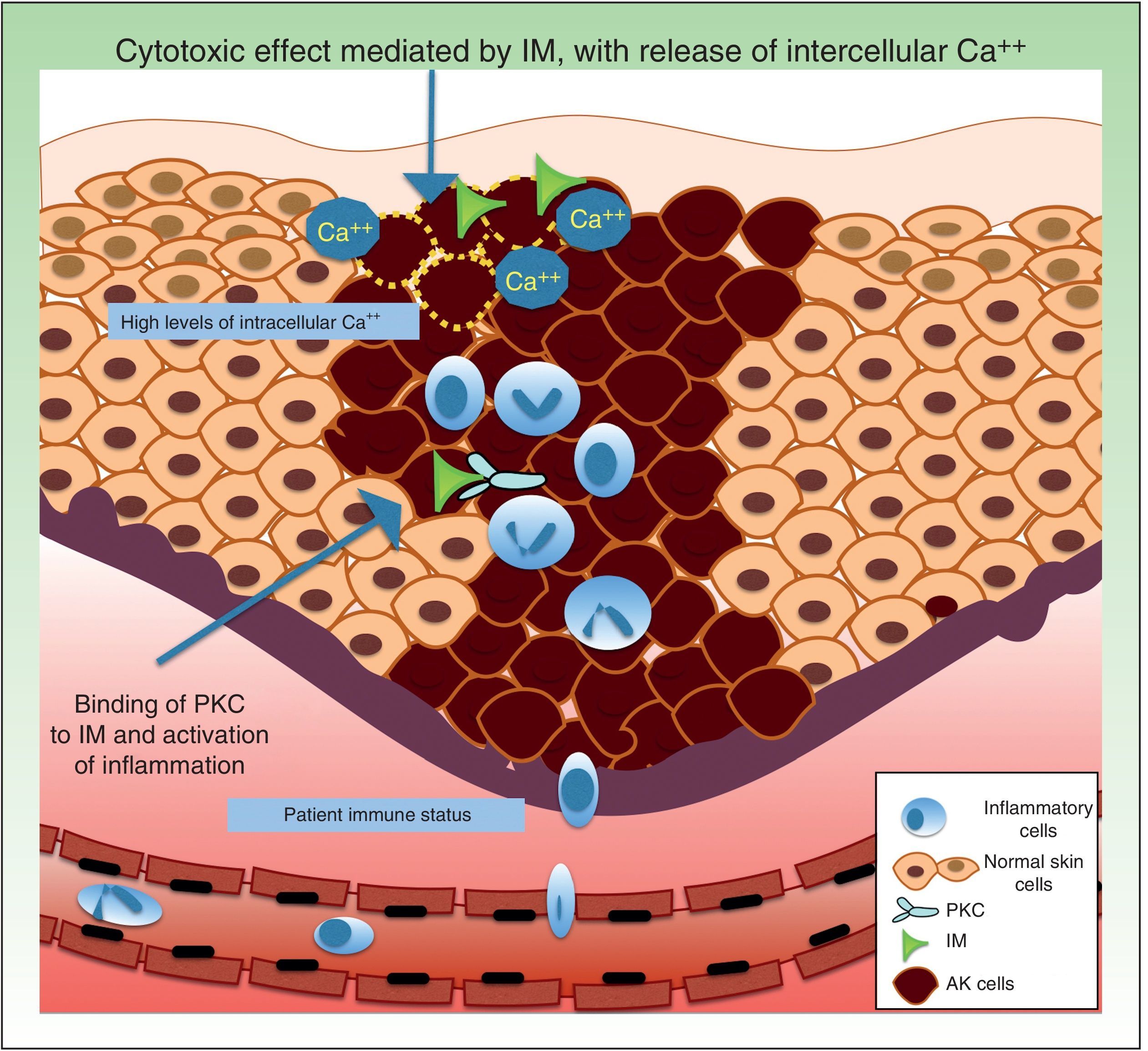
Resistance to Ingenol MebutateIngenol mebutate is a natural extract of Euphorbia peplus that has been used for many years to treat different skin conditions, such as viral warts and tumors.61 It has a dual mechanism of action. On the one hand, it rapidly induces apoptosis (in a matter of hours) by necrosis of dysplastic keratinocytes through mitochondrial damage and plasma membrane disruption,62 and on the other hand, several days later, it triggers an inflammatory response through protein kinase C δ (PKC), with the production of proinflammatory cytokines and tumor-specific antibodies that cause neutrophil-mediated antibody-dependent cellular cytoxicity (Fig. 4).63

Topical ingenol mebutate gel is approved for the treatment of AK in 2 concentrations: 0.015% applied once daily for 3 days for lesions on the head and 0.05% applied once daily for just 2 days for lesions on the trunk.64 The gel has also been used to treat other cutaneous disorders such as BCC,6 Bowen disease,7 giant porokeratosis (1 case),65 anogenital warts,66 and even recurrent melanoma in situ.67
In the case of AK, ingenol mebutate has resulted in complete response rates of 42.2% for lesions on the face and neck and 34.1% for lesions on the trunk and extremities.68
Regarding factors that might influence resistance to the acute cytotoxic effects of ingenol mebutate, it has been postulated that this substance might trigger the release of calcium from the endoplasmic reticulum rather than an influx of calcium from outside the cell. Differentiated human keratinocytes have high calcium levels, and are significantly less sensitive to ingenol mebutate–mediated cell death than undifferentiated, proliferating keratinocytes with lower calcium content.62,69
Neutrophil recruitment might also have a role in resistance to ingenol mebutate. Preclinical studies have investigated the inflammatory effect of ingenol mebutate in neutrophil-depleted Foxn1nu mice (nude mice with an autosomal recessive mutation in the FOXN1 [forkhead box N1] gene associated with T-cell immunodeficiency, alopecia, and onychodystrophy) and in CD18-deficient mice (mice with deficient leukocyte cell adhesion molecule expression).70 Both groups of mice were injected with cells from the UV-induced murine SCC line LK2, and a significant increase in tumor relapse rates (>70%) was observed after several weeks in the absence of neutrophil-mediated killing of residual tumor cells. The authors concluded that an individual's immune status could contribute to resistance to ingenol mebutate treatment.70
Key point: An immune state characterized by T cell–deficiency, polymorphic neutrophil deficiency, and other factors such as intracellular calcium levels could influence sensitivity to treatment with ingenol mebutate.
ConclusionsIn recent years, we have witnessed an increase in the number of topical treatments available for NMSC, largely due to the introduction of a new class of drugs known as topical immunomodulators. The use of these immunomodulators has given rise to studies of resistance mechanisms showing that resistance could depend on the patient's immune status and on the biochemical and molecular features of the tumor cells.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Gracia-Cazaña T, González S, Gilaberte Y. Resistencias al tratamiento no quirúrgico en cáncer cutáneo no melanoma. Parte I: tratamientos tópicos. Actas Dermosifiliogr. 2016;107:730–739.



