







La relación entre el fibroxantoma atípico (FXA) y el sarcoma pleomórfico dérmico (SPD) ha sido confusa y objeto de debate a lo largo de los años en la literatura científica. Son tumores que se presentan en pacientes de edad avanzada en piel fotoexpuesta, típicamente cabeza y el cuello. Están formados por una mezcla variable de células histiocitoides, fusiformes, epitelioides y gigantes multinucleadas con núcleos pleomórficos. No existen técnicas inmunohistoquímicas diagnósticas de estas entidades y su diagnóstico debe ser de exclusión. El FXA es una neoplasia dérmica, bien delimitada, con ausencia de infiltración difusa de tejido subcutáneo, necrosis tumoral o invasión linfovascular o perineural. Estando alguna de las características anteriores presente, debe hacerse el diagnóstico de SPD. En esta revisión narrativa de la literatura intentaremos determinar cuáles son las características histopatológicas precisas de ambas entidades, según las series publicadas en la literatura y aquellos aspectos que las diferencian o relacionan.
The relation between atypical fibroxanthoma and pleomorphic dermal sarcoma has led to confusion and debate in the literature. Both tumors present on sun-exposed skin, typically on the head and neck, in patients of advanced age. Both are comprised of a variable mix of histiocytoid, spindle, epithelioid, and/or giant multinucleated cells with pleomorphic nuclei. No immunohistochemical diagnostic techniques have emerged to distinguish these tumors. Diagnosis is by exclusion. Histologically, atypical fibroxanthoma is seen as a well-circumscribed dermal nodule but there will be no evidence of extensive subcutaneous invasion, tumor necrosis, or lymphovascular or perineural invasion. Therefore, if any of the aforementioned features is present, the diagnosis would be pleomorphic dermal sarcoma. This narrative review of the literature aims to identify the distinguishing and overlapping histopathologic features of these 2 tumors as they have been described in case series.
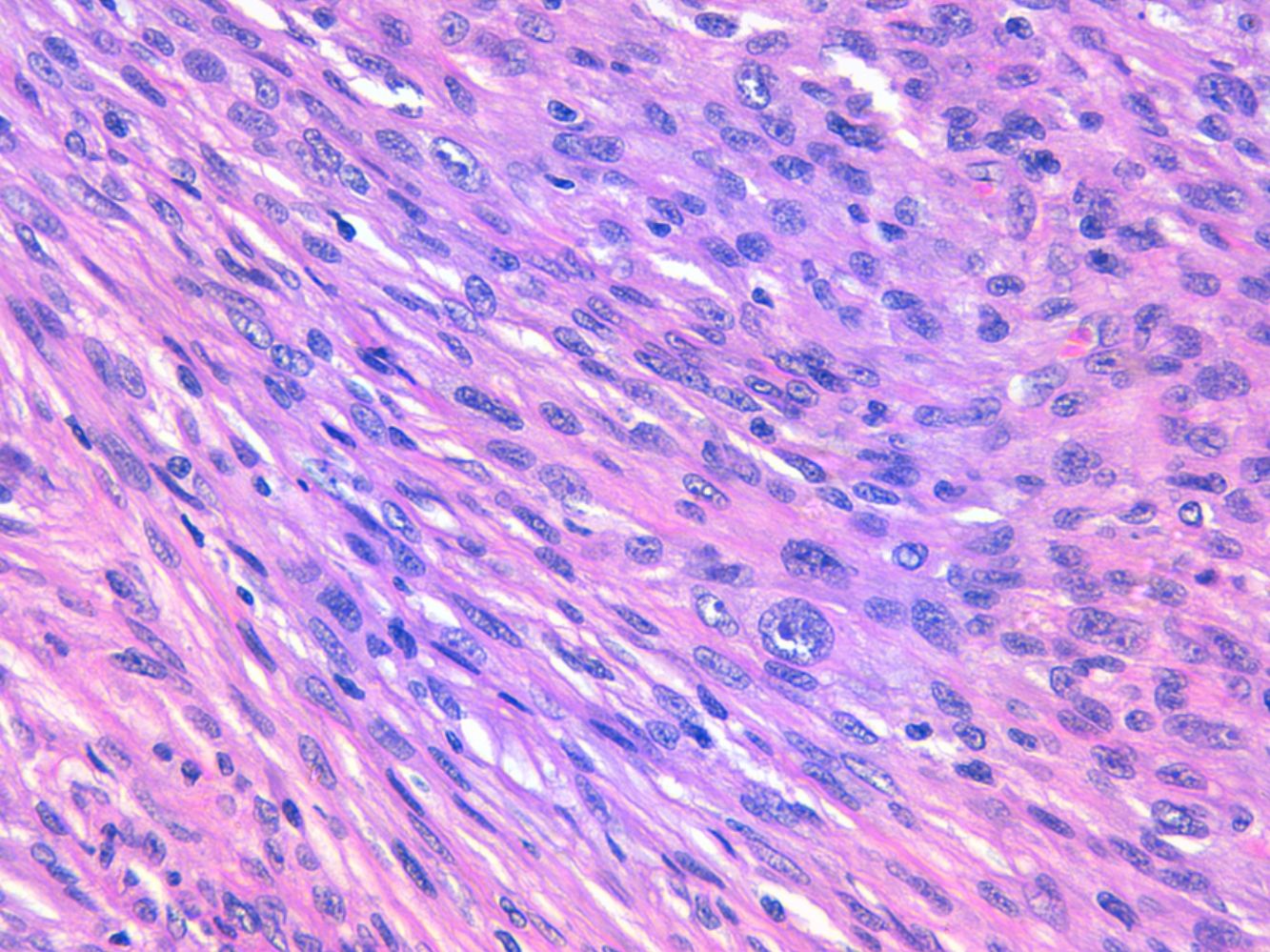
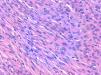
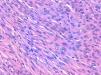
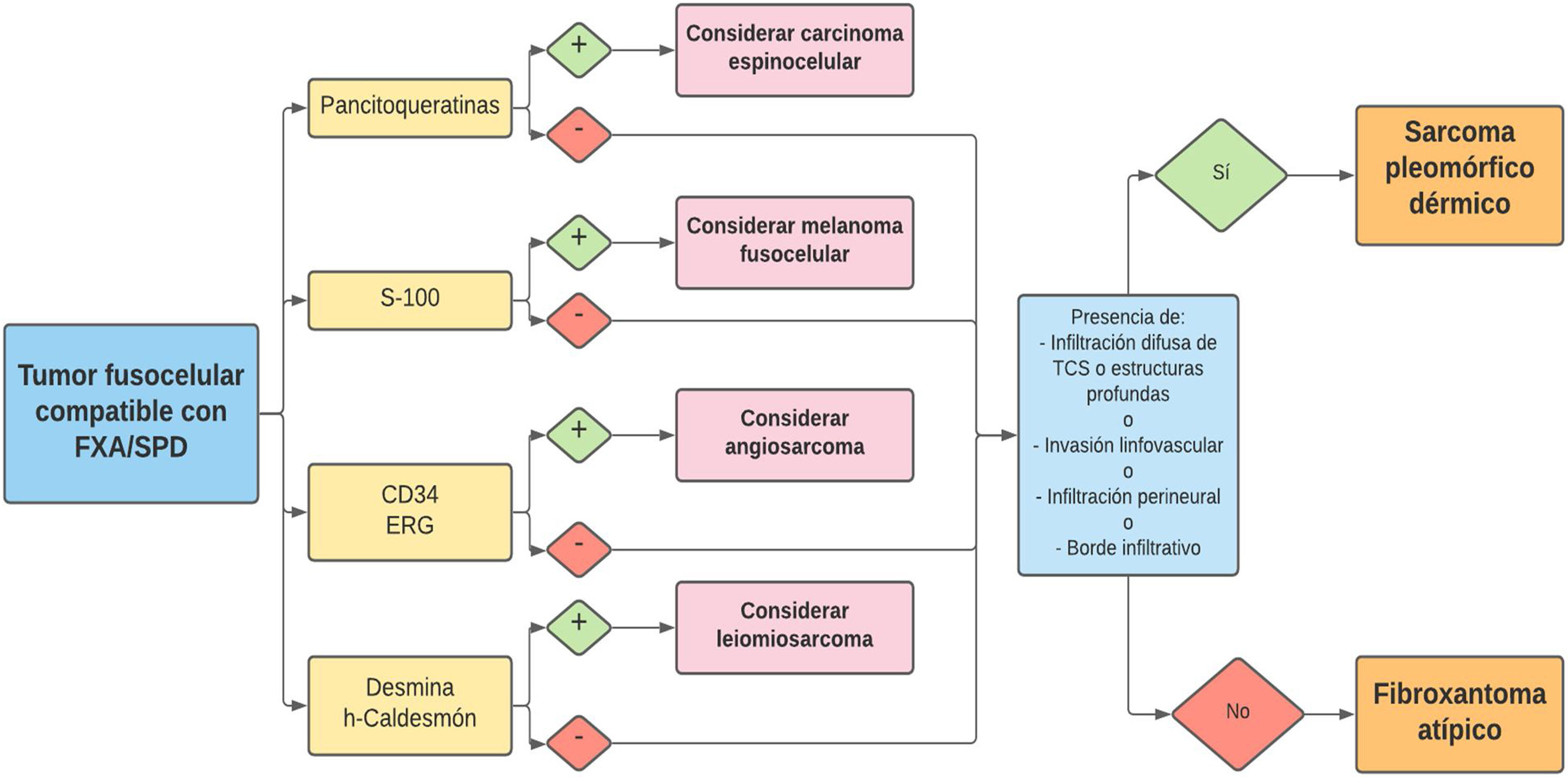
La relación entre el fibroxantoma atípico (FXA) y el sarcoma pleomórfico dérmico (SPD) ha sido confusa y objeto de debate a lo largo de los años en la literatura científica1. Son tumores que se presentan en pacientes de edad avanzada, predominantemente en varones, en piel fotoexpuesta y en cabeza y el cuello de forma mayoritaria, frecuentemente ulcerados (fig. 1)2. Están formados por una mezcla variable de células histiocitoides, fusiformes, epitelioides y gigantes multinucleadas con núcleos pleomórficos (fig. 2), muchas veces con abundantes mitosis aberrantes. No existen técnicas inmunohistoquímicas diagnósticas de estas entidades y su diagnóstico debe ser de exclusión1.
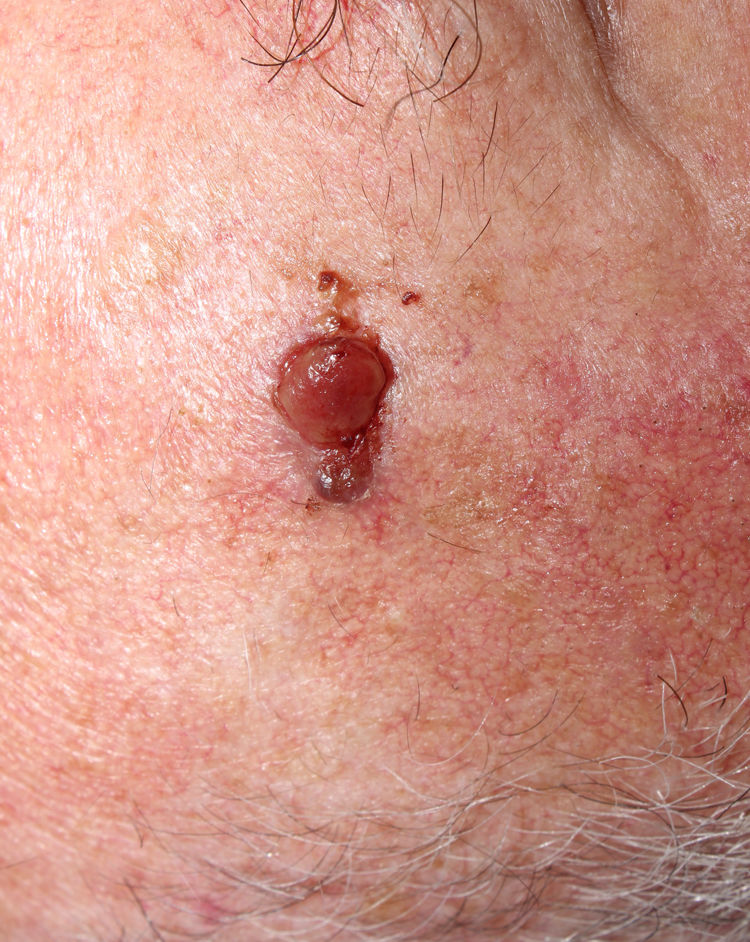
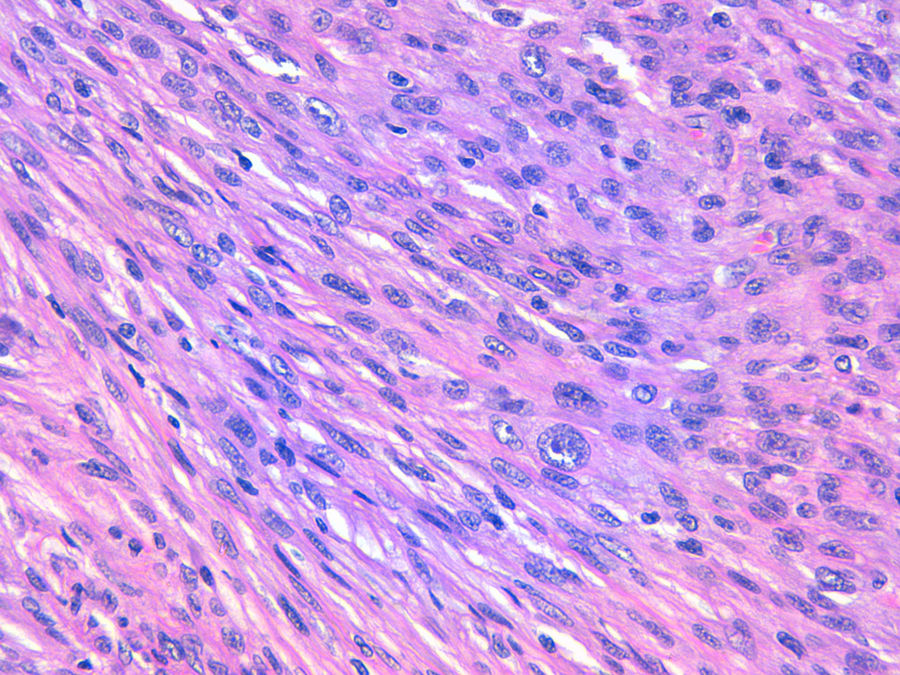
Son tumores poco frecuentes y su incidencia es desconocida, aunque la incidencia del FXA en un área sanitaria española ha sido recientemente estimada en la literatura como de 0,59 casos/100.000 habitantes3, lo que contrasta con la incidencia de otros tumores en nuestro país, como el carcinoma basocelular (113,05/100.000 habitantes), el carcinoma epidermoide (38,16/100.000) o el melanoma (8,76/100.000 habitantes)4. No se conocen cifras de incidencia del SPD.
Su etiología, en la mayoría de los casos, está relacionada con la exposición solar crónica. La implicación de la luz ultravioleta en su etiopatogenia está apoyada por la demostración de mutaciones de p535.
La definición histopatológica actual del FXA incluye las siguientes características1:
- -
Neoplasia dérmica, bien delimitada, compuesta por una mezcla de células histiocitoides, fusiformes, epitelioides y gigantes multinucleadas, con núcleos pleomórficos.
- -
Crecimiento exofítico, nodular o polipoide (fig. 3a).
- -
Ausencia de infiltración difusa del tejido celular subcutáneo, necrosis tumoral o invasión linfovascular o perineural.
- -
Diagnóstico de exclusión basado en un análisis de la pieza completa de resección, y tras descartar mediante un panel inmunohistoquímico adecuado sus principales diagnósticos diferenciales: carcinoma espinocelular sarcomatoide o fusocelular, melanoma fusocelular, leiomiosarcoma pobremente diferenciado y angiosarcoma.
- -
Puede existir collarete epidérmico.
- -
El borde profundo suele ser predominantemente expansivo.
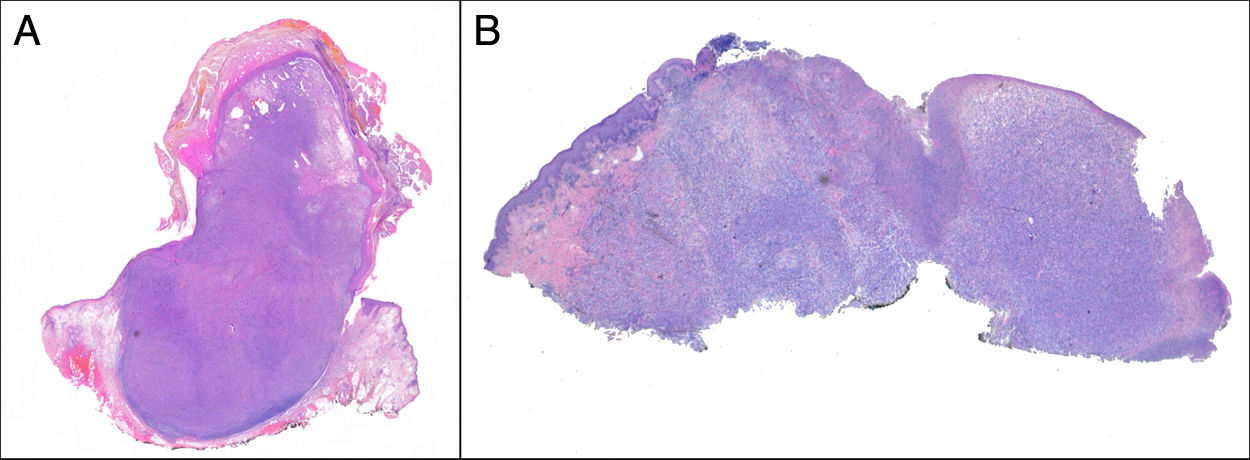
El SPD tiene características histopatológicas similares al FXA, pero con infiltración difusa del tejido celular subcutáneo o estructuras profundas, necrosis tumoral o invasión linfovascular o perineural6 (fig. 3b). Cuando se aplican de forma estricta estos criterios diagnósticos, el comportamiento clínico del FXA se considera benigno, con recidiva local infrecuente. Aquellos FXA publicados en la literatura con desarrollo de metástasis, probablemente se traten de otros tumores incorrectamente diagnosticados. Los términos «sarcoma pleomórfico indiferenciado de la piel» e «histiocitoma fibroso maligno superficial y profundo» están mal documentados y no se utilizan en la actualidad para referirse a estos tumores2.
A pesar de que FXA y SPD son tumores estudiados con detalle, existe todavía en la literatura un debate acerca de la relación entre estas dos entidades y existen cuestiones sin resolver, como si un FXA podría convertirse en SPD de forma tiempo-dependiente, formando parte ambos de la misma entidad y cuyo pronóstico depende de la profundidad de la neoplasia1. En esta revisión de la literatura intentaremos determinar cuáles son las características histopatológicas precisas de ambas entidades, según las series publicadas en la literatura y aquellos aspectos que las relacionan.
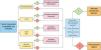
Material y métodosPara la elaboración de esta revisión, se realizó una búsqueda bibliográfica en las bases de datos PubMed (Medline), Cochrane Library, Embase, SciELO y Trip Database con los siguientes términos: «atypical fibroxanthoma» AND «pleomorphic dermal sarcoma», el 4 de septiembre de 2020. La búsqueda inicial arrojó 126 resultados (53 tras eliminar duplicados). Se realizó una lectura y criba de los resúmenes, según los siguientes criterios:
- -
Criterios de inclusión de los estudios en la revisión:
- ∘
Series de casos publicadas en revistas revisadas por pares en los últimos 10 años, que incluyan FXA y/o SPD, teniendo en cuenta la existencia de ambos tumores en los términos actualmente utilizados. También aquellos que traten la relación entre estos dos tumores, según alguna característica determinada.
- -
Criterios de exclusión:
- ∘
No relacionados con el objeto de la revisión (cinco publicaciones).
- ∘
Casos clínicos aislados (11 publicaciones).
- ∘
Revisiones narrativas sobre fibroxantoma atípico y/o sarcoma pleomórfico dérmico (14 publicaciones).
En total, 24 estudios fueron seleccionados para la lectura completa, de los cuales 22 fueron finalmente incluidos en la presente revisión (fig. 4). El motivo de exclusión de estos dos últimos estudios fue la agrupación de características de los casos de FXA y SPD, sin aplicación de criterios diagnósticos para su diferenciación.
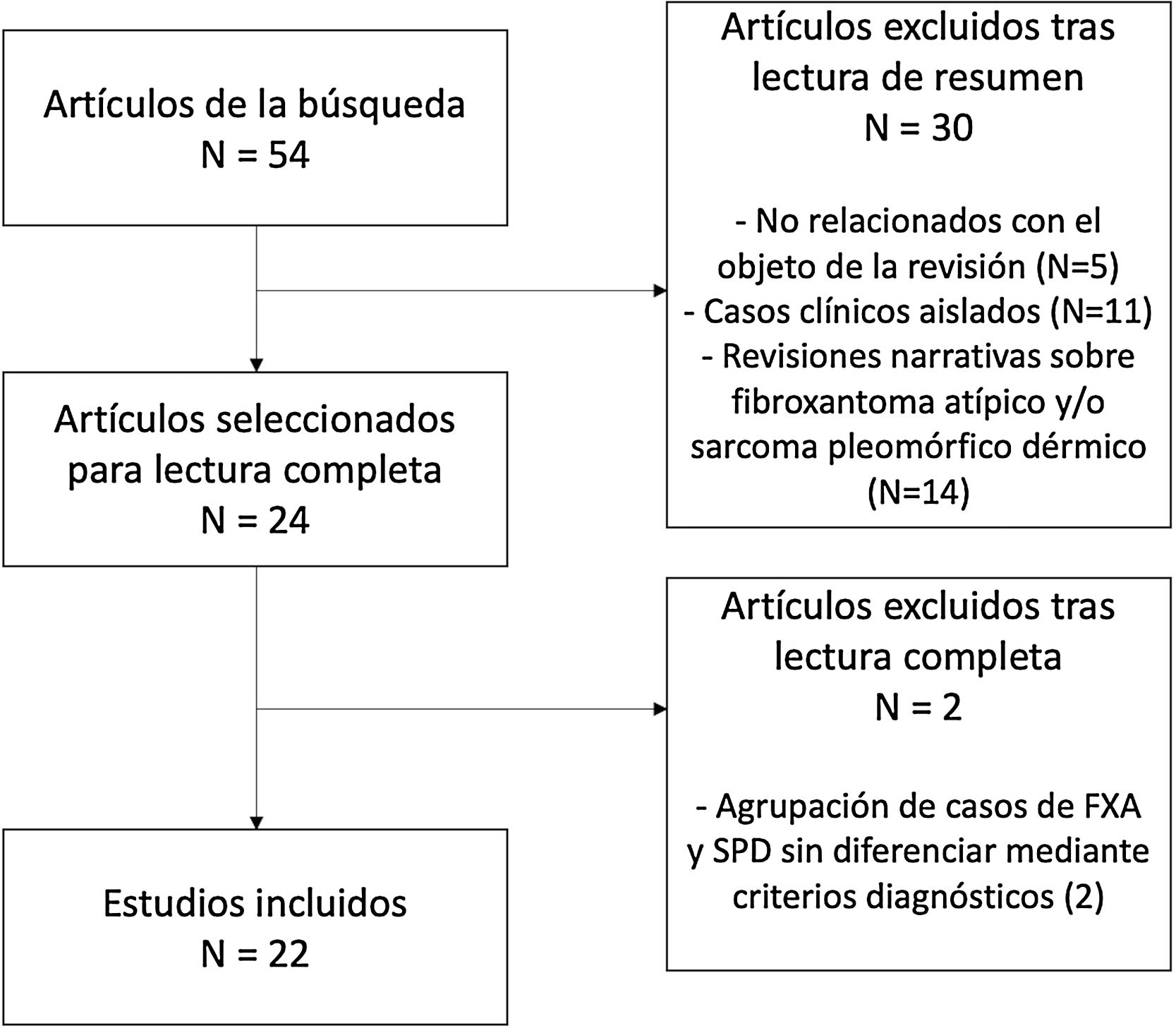
De las series incluidas, se recogieron los siguientes datos: pacientes y tumores, localización, criterios diagnósticos utilizados, patrón de infiltración, tamaño, índice mitósico, ulceración, necrosis, infiltración perineural, invasión linfovascular, borde profundo afectado, variantes, inmunohistoquímica (marcadores negativos), pronóstico y relación entre tumores.
ResultadosLos resultados de las series incluidas de FXA3,7–10, SPD2,6 y mixtas11–25, se resumen en la tabla 1.
Series de casos incluidas en la revisión
| Estudio | Pacientes y tumores | Localización | Criterios diagnósticos utilizados | Patrón de infiltración | Tamaño tumoral | Índice mitósico | Ulceración presente | NTIPNILV | Borde profundo afectado | Variantes/Citología | Inmunohistoquímica (marcadores negativos) | Pronóstico | Relación entre tumores |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Miller et al. (2012)2 | 32 SPD | Cabeza (96,8%)Antebrazo (3,2%) | Invasión de TCS profunda (al menos áreas profundas de tejido subcutáneo, músculo, fascia o galea) y/o NT, ILV o IPNS100, CD34 y desmina negativos | Infiltrativo (59,3%)Expansivo (37,5%)No determinado(3,1%) | Mediana 25 mm (rango 7 a 60 mm) | 19,5 mitosis/10 CGA (rango 6 a 51) | 25 (78%) | NT (53%)IPN (28,1%)ILV (25%) | 34,3% | Clásica 40,6%Predominio fusocelular 59,3%Cambios mixoides 18,7%Respuesta estromal desmoplásica 21,8%Pseudoangiomatoso/hemorrágico 9,3%Patrones estoriformes 6,2%Cambios queloideos 6,2%Espacios telangiectásicos 3,1%Similar a osteoclastos 3,1% | AE1/3MNF116cam5.2CK5/634bE12CK14S100HMB-45Desminah-CaldesmónCD34 | Mediana seguimiento 24 meses (disponible para 29 pacientes)RL 28%MT 10% | NA |
| Thum et al. (2013)11 | 11 FXA | CyC (100%) | Centrados en dermisNo evidencia de precursor epidérmico | Expansivo (número de casos NE) | Mediana 12 mm (rango 7 a 23) | «Alto» (sin especificar tipo de tumor) | 10 casos (sin especificar tipo de tumor) | Ausentes | NE (al menos 1) | Pseudoangiomatoso/hemorrágico (100%) | AE1/AE3MNF116 CK14 CK5/6 S100 melan AHMB-45 Desmina p63 | Mediana seguimiento 43,1 meses (10 pacientes)RL 0%MT 0% | Ambos tumores negativos para ERG y CD34 |
| 3 SPD* | Invasión de la fascia subyacente | NE | Mediana 35 mm (rango 20 a 40) | NE (al menos 1) | |||||||||
| Hollmig et al. (2013)26 | 8 SPD | CyC (100%) | Tumor infiltrando TCS | NE | NE | NE | NE | NE | NE (4 casos con biopsia «insuficiente para su evaluación») | NE | NE | Media seguimiento 19,5 mesesRL 62,5%MT 25% | Características inmunohistoquímicas similaresLN-2 no es buen marcador diagnóstico ni pronóstico en estos tumores |
| 14 FXA | Tumor limitado a la dermis | NE | Media seguimiento 43 mesesRL 14,2%MT 14,2% | ||||||||||
| Zschoche et al. (2014)18 | 25 FXA | CyC (100%) | Patrón de crecimiento exofíticoNo invasión de TCS | NE | NE | NE | NE | NE | NE | NE | NE | NE | Ambos tumores presentan una densidad similar de vasos linfáticos intratumoralesNo hay diferencias significativas entre FXA y SPD cuando se comparan los 3 subgrupos de linfáticos |
| 22 SPD | CyC (95,5%)Brazo (4,5%) | Infiltración de TCSTumores en dermis profunda sin patrón de crecimiento exofítico, aun sin infiltración de TCS | |||||||||||
| Nonaka, Bishop (2014)19 | 19 FXA | CyC (100%) | Tumor limitado a la dermis | NE (datos agrupados de FXA y SPD) | NE (datos agrupados de FXA y SPD) | NE (datos agrupados de FXA y SPD) | NE (datos agrupados de FXA y SPD) | NE (datos agrupados de FXA y SPD) | NE | NE | AE1/AE3MNF11634bE12CK5/6CK14p63 | NE (datos agrupados de FXA y SPD) | No explorada porque se expresan los datos de forma agrupada |
| 34 SPD | Tumor se extiende a TCS | ||||||||||||
| Griewank et al. (2014)20 | 27 FXA | CyC (96,2%)Desconocido (3,8%) | Ausencia de invasión de TCS, NT, IPN e ILV | Expansivo (100%) | Mediana 9 mm (rango 4-30) | Mediana 20 mitosis/mm2 (rango 7-53) | 44,4% | Ausentes | NE | NE | MNF116AE1/ AE3CD31 (focal en 1 caso)CD34S100Desmina | NE | Las mutaciones en el promotor de TERT están presentes en el 93% de los FXA y en el 76% de los SPD. El número de mutaciones CC > TT sugiere un rol patogénico de la luz UV en ambos tumores |
| 34 SPD | CyC (94,2%)Desconocido (5,8%) | Presencia de invasión de TCS, NT, IPN o ILV | Infiltrativo (50%)Expansivo (47%)No evaluable (2,9%) | Mediana 22 mm (rango 6-60) | Mediana 19 mitosis/mm2 (rango 5-51) | 76,4% | NT (55,8%)IPN (26,4%)ILV(26,4%) | ||||||
| Harding-Jackson et al. (2015)7 | 15 FXA | CyC (86,6%)Desconocido (13,3%) | Circunscrito con borde expasivoAusencia de NT, IPN, ILV o extensión profunda a TCS | Expansivo (100%) | NE | Rango 5-38 por 10 CGA | 60% | Ausentes | NE | NE | DesminaCalponinah-CaldesmónS100P63CK amplio espectroCD31CD34 | Al menos 2 años de seguimientoRL 6,6% (1 caso)MT 0% | NA |
| Wang et al. (2015)8 | 11 FXA | CyC (100%) | Incluyen tumores con invasión de TCS, NT y con IPN | Infiltrativo (90,9%)No disponible (9,1%) | Mediana 8 mm (rango 3-18) | Mediana 20 mitosis por 10 CGA (rango 10-55) | 27,27% | NT 18,18%IPN 45,45%No ILV | NE | Fusocelular (6/11)Fusocelular con células gigantes aisladas (2/11)Fusocelular y epitelioide (1/11)Fusocelular y pleomórfica (1/11)Pleomórfico (1/11) | ?NE | Selección de 9 casos con metástasis de 152 (5,9%) + 2 seleccionados de otro centro2 casos sin afectación de TCS metastatizaron | NA |
| Tardío et al. (2016)21 | 18 SPD | CyC (100%) | Invasión de tejido subcutáneo profundo y/o NT y/o IPN y/o ILV | Expansivo (17%)Infiltrativo (83%) | Mediana 15 mm (rango 7-70) | Mediana de 22 mitosis por 5 mm2 (rango 2-126) | 55,5% | NT 17%ILV 17% | 22,2% | Fusocelular fascicularCélulas epitelioides gigantes multinucleadas (2/18)Células neoplásicas con núcleo vesicular moderadamente pleomórficas y nucleolo prominente | CitoqueratinasS100DesminaCD34 | Seguimiento con mediana de 33 meses (disponible en 15/18)RL 20%MT 20% | Mismas características histopatológicas e inmunohistoquímicas, excepto la carencia de invasión de TCS profunda, NT o ILV en los FXAÚnicamente diferencias en el pronóstico |
| 45 FXA con seguimiento de más de 12 meses (para comparación) | CyC (100%) | Ausencia de invasión de tejido subcutáneo profundo, NT, IPN o ILV | NE | Mediana 11mm (rango 4-30) | NE | NE | Ausentes | NE | NE | NE(«las mismas») | Seguimiento de más de 12 meses (mediana de 48 meses)RL 2,2% (1 caso)No MT | ||
| Helbig et al. (2016)22 | 5 FXA | Muslo (20%)CyC (80%) | Limitado a la dermis | NE | NE | NE | NE | NE | NE | NE | Al menos una citoqueratina (CK 5/6 o pancitoqueratina)Dos marcadores melanocíticos (S100, melan A o HMB-45)Marcadores vasculares (CD31, CD34 o podoplanina) | NE | Todos los FXA y SPD presentaron expresión de oncogenes similar (sobreexpresión de TP53, CCND1, CDK4)Un caso con FXA y SPD mostró perfiles mutacionales de TP53 y PIK3CA idénticos en ambos tumores |
| 6 SPD (un paciente con FXA y SPD separados por 3 años) | CyC (100%) | Infiltración de TCS | |||||||||||
| Helbig et al. (2017)23 | 5 FXA | CyC (80%)Desconocido (20%) | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | Detectadas amplificaciones y deleciones en 6 de 27 SPD; no se detectaron en los FXA |
| 26 SPD | CyC (96,1%)Hombro (3,9%) | ||||||||||||
| Griewank et al. (2018)24 | 41 FXA | CyC (90,2%)Brazo (4,8%)Desconocido (4,8%) | Establecidos tras el análisisAsociación ES de SPD con NT, invasión de TCS, invasión de fascia/músculo, borde infiltrativo e ILV | Infiltrativo (2,4%)Expansivo (73,1%)No evaluable (24,3%) | Mediana 8 mm (rango 4-30) | Mediana de 17 mitosis/mm2 (3-52) | 34,1%No determinado 17,1% | NT 2,4% (1 caso incluido)No IPNNo ILV | NE | NE | Pancitoqueratina (MNF116, AE1/AE3) CD31 o CD34S100DesminaActina músculo lisomelan A (1 caso expresión aberrante) | NE | FXA y SPD presentan mutaciones recurrentes en FAT1, NOTCH1/2, CDKN2A, TP53 y el promotor de TERT |
| 40 SPD** | CyC (92,5%)Brazo (2,5%)Desconocido (5%) | Infiltrativo (40%)Expansivo (25%)No evaluable (35%) | Mediana 20 (rango 4-60) | Mediana de 21 mitosis/mm2 (rango 5-44) | 55%No determinado 25% | NT 40%IPN 22,5%ILV 20% | |||||||
| Helbig et al. (2018)25 | 25 FXA | CyC (96%)Pierna (4%) | No invasión de TCS | NE | NE | NE | NE | NE | NE | NE | AE1/AE3CK5/6p40SOX10ERGNecesario CD 10 + | NE(solo disponible para los dos pacientes con expression de MiTF: sin progresión tumoral) | Expresión de MiTF en 1/25 FXA y 1/25 SPD; expresión de α-SMA en 40% de FXA y 36% de SPD |
| 25 SPD*** | CyC (92%)Hombro (8%) | Infiltración de TCS, fascia o músculo | |||||||||||
| Gaiser et al. (2018)13 | 51 FXA | CyC (100%) | NE | NE | NE | NE | NE | NE | NE | NE | NE | RL 1,9% (1 caso)No MT | La amplificación de MYC no es un factor genético determinante en el proceso de génesis tumoral de FXA o SPD |
| 24 SPD | CyC (91,7%)Hombro (8,3%) | NE | |||||||||||
| Koelsche et al. (2019)14 | 17 FXA | CyC (100%) | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | El perfil de metilación de ADN no distingue FXA y SPDPérdidas de 9p y 13q y ganancias de 8q de frecuencia similarDeleción homocigota de CDKN2A más frecuente en SPD que en FXA |
| 15 SPD | CyC (93,3%)Desconocido (6,6%) | ||||||||||||
| Müller et al. (2019)9 | 41 FXA(40 pacientes) | CyC (90,3%)Extremidades (9,7%) | NEIncluyen pacientes con nivel de Clark V y necrosis pero sin ILV | NE | Media de 5,7 cm2 (rango 0,06 y 40) | Mediana 4,8 mitosis/10 CGA (rango 1-50,4) | 68,2% | NT 14,6%No IPNNo ILV | NE | NE | NE | RL 7,3%No MT | NAAlta expresión de SEC62 en tumores con necrosis, tamaño > 5 cm2, altos niveles de Clark |
| Nassios et al. (2019)15 | 4 FXA | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | Expresión similar de GPR4, TDAG8, OGR1 y G2A en FXA y SPD estudiados |
| 5 SPD | |||||||||||||
| Miller et al. (2020)16 | 21 FXA | CyC 90,5%Rodilla 4,7%Pecho 4,7% | Proliferación fusocelular atípica en piel fotodañada con mínima o ninguna infiltración de TCS, sin NT, ILV, IPN o diferenciación epitelial/melanocítica/vascular/músculo liso por morfología o inmunotinciones | NE | Mediana 0,9 mm (rango 0,2-2,8) | NE | NE | Ausentes | NE | NE | CitoqueratinasS100Otros (ver tabla suplementaria en artículo original) | Seguimiento en 7 pacientesNo RL ni MT | Detectadas mutaciones en PIK3CA (no en SPD), deleciones en CDKN2A fueron más frecuentes en SPD que en FXA, no se observaron mutaciones en CDKN2A en SPD y sí en FXA, más mutaciones en el promotor de TERT de los SPD en relación a FXA |
| 17 SPD | CyC 88,2%Muslo 5,8%Supraclavicular5,8% | Presencia de infiltración subcutánea profunda, necrosis, invasión linfovascular y/o invasión perineural | NE | NE | NE | NE | NE | NE | Seguimiento en 2 pacientes (ambos RL) | ||||
| Lonie et al. (2020)6 | 27 SPD | CyC (96,3%)Torso (3,7%) | Citoqueratinas, S100 y CD34 negativos, sin especificar otros criterios histopatológicos | NE | NE | «Numerosas» 35%«Frecuentes» 33,3% | NE | NE | NE | NE | CitoqueratinasS100CD34 | Mediana de seguimiento de 46,4 mesesRL 7,3%MT 3,7% (1 caso) | NA |
| Ricci et al. (2020)17 | 5 FXA | CyC (100%) | NE | NE | NE | NE | NE | NE | NE | NE | Citoqueratinas | NE | La catepsina-K mostró positividad moderada y difusa en todos los casos de FXA y SPD |
| 4 SPD | CyC (50%)Tronco (50%) | ||||||||||||
| Bitel et al. (2020)10 | 105 FXA(85 pacientes) | CyC (97,9%)Hombros (2,1%) | NEIncluyen tumores infiltrando músculo/fascia/cartílago | NE | 15 mm +/- 3 mm | «Alta» 63%«Intermedia» 17,8%«Baja» 19,2% | 47,6% | NE | NE | Fusocelular 70%Mixto 30% | NE | Seguimiento de 30 meses de media en 36 pacientesRL 22,9%No MT | NAIdentifican como factor de riesgo de recidiva la infiltración de estructuras profundas |
| Iglesias-Pena et al. (2020)3 | 62 FXA | CyC (96,8%)Tronco (3,2%) | Infiltración de estructuras profundasPanel inmunohistoquímico compatibleAusencia de NT, IPN e ILV | NE | Media 12,3 mm (rango 3-40) | Mediana de 7,09 mitosis/10 CGA (rango 0-31) | 50% | Ausentes | 8,1% | Clásica 88,7%Fusocelular no pleomórfica 4,8%Hemosiderótica 4,8%Queloidea 4,8% | AE1-AE3CD34HMB45DesminaEMAS100 (1 focal) | Seguimiento mediana de 34 mesesRL 6,5% (4 casos)No MT | NA |
CGA: campos de gran aumento; CyC: cabeza y cuello; ES: estadísticamente significativa; FXA: fibroxantoma atípico; ILV: invasión linfovascular; IPN: infiltración perineural; MT: metástasis; NA: no aplica; NE: no especificado; NT: necrosis tumoral; SPD: sarcoma pleomórfico dérmico; RL: recidiva local; TCS: tejido celular subcutáneo.
Las series en general son cortas, de pocos pacientes y raramente superando los 30 tumores de cada tipo. La localización predominante fue la cabeza y el cuello, con el 100% de los tumores en dicha localización, en muchas series.
Diferencias entre FXA y SPDEl FXA y el SPD se han diferenciado de forma mayoritaria a lo largo de las series, mediante criterios histopatológicos. En la serie de Griewank et al. de 2014, la diferenciación entre FXA y SPD es realizada por dos dermatopatólogos y en el análisis posterior encuentran que necrosis, invasión de tejido subcutáneo, invasión de fascia/músculo, borde infiltrativo e invasión linfovascular son criterios asociados de forma estadísticamente significativa a SPD24.
Los criterios diagnósticos utilizados para la distinción entre FXA y SPD no son homogéneos en los estudios, y en muchas ocasiones no están claramente especificados. Los tumores con necrosis tumoral, invasión linfovascular, invasión perineural o invasión de fascia/músculo fueron generalmente clasificados como SPD, aunque algunas series consideraron como FXA tumores con alguna de estas características8–10,24.
Mención especial merece la infiltración del tejido celular subcutáneo para la distinción de FXA y SPD. Mientras que, en algunas series12,18–20,22,25, la simple infiltración de tejido celular subcutáneo se considera automáticamente criterio de SPD, en otras2,7,11,21, se establece que dicha infiltración debe ser «profunda», en algunos casos sin especificar cuánta debe ser esa profundidad. En una serie incluso se consideraron SPD los tumores dérmicos que no infiltraban tejido subcutáneo, por el hecho de no presentar un patrón de crecimiento exofítico18 (tabla 2).
Diferencias en la consideración de FXA y SPD, según la infiltración de tejido subcutáneo en algunas de las series de casos incluidas
| Simple infiltración de TCS como criterio de SPD | Necesaria infiltración profunda de TCS para considerar SPD |
|---|---|
| Hollmig et al. (2013)26Zschoche et al. (2014)18*Nonaka, Bishop (2014)19Griewank et al. (2014)20Helbig et al. (2016)22Helbig et al. (2018)25 | Miller et al. (2012)2Thum et al. (2013)11Harding-Jackson et al. (2015)7Tardío et al. (2016)21 |
Muchas series no especifican el patrón de infiltración de los tumores. Cuando lo especifican, la mayoría de los FXA presentan un patrón expansivo; mientras que los SPD presentan patrones mayoritariamente infiltrativos2,7,20,21,24. Es de destacar el porcentaje de FXA con patrón infiltrativo en la serie de Wang (90,9%), de 11 FXA metastásicos, que incluyen tumores con invasión de tejido subcutáneo extensa, necrosis tumoral e infiltración perineural, criterios que deberían reclasificar dichos tumores como SPD8.
El tamaño tumoral11,20,24 y el índice mitósico20,24, cuando están recogidos, son claramente superiores en los SPD frente a los FXA. Del mismo modo, la ulceración es superior en los SPD en los estudios en los que esta se especifica de forma diferenciada20,24.
En la serie de FXA de Müller et al.9 se encontró una expresión de SEC62 incrementada estadísticamente significativa en tumores con necrosis y una tendencia a mayor expresión en tumores con altos niveles de Clark y tamaño mayor de 5 cm2.
A nivel genético, debemos destacar la serie de Miller et al. (2020), en la que detectan mutaciones en PIK3CA (y no en SPD), deleciones en CDKN2A más frecuentes en SPD con respecto a FXA, mutaciones en CDKN2a en FXA que no se observaron en SPD y un aumento de mutaciones en el promotor de TERT de los SPD en relación a FXA16.
Finalmente, en cuanto al pronóstico, las diferencias entre FXA y SPD son notables. En las series que incluyen ambos tipos de tumores21,26, la recidiva local y las metástasis son claramente superiores en los SPD. Es de destacar que las series que incluyen FXA metastásicos, en las que especifican las características histopatológicas8,10, incluyen tumores con infiltración de estructuras profundas, necrosis tumoral o infiltración perineural (que, según los criterios actuales, tendríamos que considerar SPD).
Similitudes entre FXA y SPDEn la presente revisión podemos encontrar similitudes entre el FXA y el SPD en las diferentes series que comparan estos tumores.
A nivel inmunohistoquímico, ningún estudio demuestra la existencia de un marcador que se exprese de forma diferente entre los dos tipos de tumores26. Los marcadores inmunohistoquímicos utilizados varían entre los estudios, pero incluyen mayoritariamente algunas citoqueratinas, S100 y marcadores vasculares (CD31, CD34), aunque en algunas series no se especifican de forma clara8–10,12–15,18,23.
Las investigaciones de Zschoche et al. no mostraron diferencias en la arquitectura linfática en más de una veintena de ejemplos de cada tumor18.
En la serie de Nonaka y Bishop, el FXA y el SPD se presentan de forma agrupada como tumores sarcoma-like, sin evidencia inmunohistoquímica de diferenciación epitelial, sin signos histológicos de componente de carcinoma espinocelular19. Esto impidió valorar de forma separada las características de estos tumores en esta revisión. Curiosamente, estos autores demostraron que el pronóstico de FXA y SPD de forma agrupada era similar al de otros tumores sarcoma-like con componente epitelial, sugiriendo que al menos algunos casos de FXA/SPD podrían estar relacionados con el carcinoma espinocelular y representar los primeros una pérdida completa del fenotipo epitelial.
A nivel genético, la serie de Griewank et al. (2018) muestra que las mutaciones en el promotor de TERT está presente en el 93% de los FXA y en el 76% de los SPD. El número de mutaciones CC > TT sugiere un rol patogénico de la luz UV en ambos tumores20. En la serie de Helbig et al., se comprobó que FXA y SPD tenían una expresión de oncogenes similar, con sobreexpresión de TP53, CCND1 y CDK4, lo que lleva a los autores a reafirmar su hipótesis de que el FXA es el precursor no infiltrativo de del SPD22. En otra serie de Helbig de 2018, con 25 FXA y 25 SPD, los autores encuentran expresión de MiTF en un FXA y un SPD, así como expresión de α-SMA en el 40% de los FXA y el 36% de los SPD de la muestra25. También Gaiser et al. descartan en otra serie de pacientes que la amplificación del gen MYC sea un proceso determinante en la génesis tumoral de FXA y SPD13. Koelsche et al. demostraron que el perfil de metilación de ADN no distingue FXA de SPD; en su trabajo observaron pérdidas de 9 q y 13 q y ganancias de 8 q, en frecuencia similar en ambos tumores. La deleción homocigota del CDKN2A fue más frecuente en el SPD (6/15), con respecto al FXA (2/17), aunque con una muestra relativamente baja14.
Nassios et al. estudiaron la expresión de receptores acoplados a proteínas G sensibles a protones y encontraron una expresión similar de GPR4, TDAG8, OGR1 y G2A en los FXA y SPD estudiados15.
DiscusiónSalvo los criterios histopatológicos previamente mencionados (infiltración de estructuras profundas, necrosis tumoral, infiltración perineural e invasión linfovascular), no queda claro qué es lo que realmente diferencia al FXA del SPD. La literatura es muy confusa debido a la diferente terminología, los diferentes criterios diagnósticos utilizados por los autores y por la caracterización inmunohistoquímica insuficiente de los tumores27, especialmente en artículos antiguos28. Es fundamental realizar una evaluación exhaustiva de las piezas de resección cuando se sospechan estos tumores, pues el diagnóstico final tiene implicaciones en el manejo posterior de los pacientes29.
Existen variantes de FXA descritas en la literatura y su conocimiento es importante para evitar diagnósticos erróneos. Estas incluyen el FXA de células fusiformes no pleomórfico, el FXA de células claras, el FXA hemosiderótico, FXA mixoide, FXA rico en células gigantes similares a osteoclastos, FXA queloideo y FXA de células granulares1. También se han descrito estos cambios en algunos SPD, bien afectando a zonas del tumor o a la lesión entera2.
En los fibroxantomas atípicos se puede observar un infiltrado inflamatorio crónico, sobre todo en la periferia del tumor y es la norma la presencia de una intensa elastosis solar de la dermis adyacente5.
Los marcadores inmunohistoquímicos que ayudan a diferenciar FXA de otros tumores incluyen CD99, S-100, CD34, citoqueratinas, desmina, CD10, vimentina, HMB-45, CD68 y p63, entre otros9. El diagnóstico diferencial con el angiosarcoma de estos tumores es importante y CD34 y ERG se han demostrado útiles en este contexto, especialmente en los FXA/SPD de patrón pseudoangiomatoso/hemorrágico11 (fig. 5). Hasta el momento presente, los intentos por diferenciar FXA y SPD con marcadores inmunohistoquímicos (CD99, LN-2) no han resultado útiles12,30.
La positividad intensa con CD10 en el FXA y el SPD puede apoyar el diagnóstico. No obstante, es preciso aclarar que cualquier tumor con morfología fusiforme puede mostrar positividad a este marcador, incluyendo sarcomas como el mixofibrosarcoma5.
También es importante destacar que las células dendríticas presentes en las biopsias de FXA y SPD son positivas con S100, pero las células tumorales deben ser negativas para este marcador para realizar el diagnóstico5.
Destacar en este sentido la serie de Müller et al.9, en la que parece haber un incremento de expresión de SEC62 estadísticamente significativa en tumores con necrosis y una tendencia a mayor expresión en tumores con altos niveles de Clark y tamaño mayor de 5 cm2. Puesto que, aunque en el estudio no se especifican los criterios histopatológicos para el diagnóstico de FXA utilizados, el hecho de incluir tumores con necrosis, tamaño grande e infiltración subcutánea hace pensar que, según los criterios actuales, algunos tumores incluidos son en realidad SPD. Esta expresión incrementada de SEC62 en SPD frente a FXA podría ser objeto de nuevas investigaciones en el futuro, con inclusión de ambos tipos de tumores con criterios diagnósticos más estrictos.
Dado que los diagnósticos de FXA y SPD son de exclusión, los marcadores inmunohistoquímicos negativos sugestivos de otras entidades son los que se utilizan para descartarlas. No se ha establecido en la literatura un número o tipo de marcadores que necesariamente tengan que ser negativos para establecer el diagnóstico, lo cual produce que en muchas series de casos la caracterización inmunohistoquímica de los tumores sea insuficiente y puedan, en realidad, corresponderse con otras entidades31.
Cabe preguntarse si estando ante tumores tan relacionados como el FXA y el SPD tiene sentido continuar denominándolos con diferentes términos. Un melanoma in situ no tiene el mismo pronóstico que un melanoma ulcerado, con invasión linfovascular y microsatelitosis, pero continuamos refiriéndonos a ambos casos con el término «melanoma». Si hasta el momento no se han encontrado diferencias significativas a nivel celular, inmunohistoquímico, genético o molecular, tiene sentido agrupar estos tumores en el mismo espectro y denominarlos igual. Esto es lo que hicieron Winchester et al. en una serie de 319 pacientes con tumores englobados con el término «sarcoma pleomórfico indiferenciado», que incluyó de forma agrupada casos de FXA y SPD, motivo por el cual no fue incluido en esta revisión. En esta serie, la recidiva local ocurrió en 45 casos (14,1%) y metástasis a distancia, en 33 pacientes (10,3%). Tras realizar un completo análisis, estos autores concluyen que el comportamiento tumoral agresivo en estos tumores de forma agrupada depende de la invasión más allá de la grasa subcutánea, el tamaño tumoral mayor de 2 cm, la inmunosupresión y la presencia de invasión linfovascular32. Otro enfoque similar fue aplicado por Cesinaro et al. en una serie de 71 tumores compatibles con FXA o SPD agrupados y con un tiempo de seguimiento entre 17 y 125 meses –tampoco incluida en esta revisión–. En este estudio solo se evidenciaron cuatro recidivas locales y ningún caso de metástasis. La morfología predominantemente fusiforme se asoció con esta serie a invasión subcutánea y recidiva28. En la literatura ya existen autores que consideran al FXA y al SPD parte del mismo espectro tumoral, aunque continúan utilizándose estos términos para su diferenciación33.
En cuanto a la infiltración subcutánea, hemos demostrado en esta revisión que los criterios para considerarla, a efectos de clasificación de estos tumores, no son en absoluto homogéneos. Estas variaciones de criterio, a la hora de clasificar los tumores, probablemente han producido un sobrediagnóstico de FXA o de SPD, según el caso, con la consiguiente alteración de las tasas de recidiva y/o metástasis en estos tumores. A lo largo de la literatura se ha definido el FXA como una neoplasia dérmica limitada a la dermis o con «mínima» infiltración del tejido celular subcutáneo. Hasta qué limite exacto de invasión podemos llamar de forma segura al tumor FXA nunca ha quedado establecido de forma homogénea34,35. En la propia clasificación de tumores cutáneos de la OMS, se especifica inicialmente que el FXA está circunscrito en la dermis «sin invasión del tejido celular subcutáneo» para, a continuación, afirmar en el diagnóstico diferencial que «lesiones que se asemejan a FXA pero son de gran tamaño o muestran invasión sustancial del tejido celular subcutáneo o más allá, invasión perineural, invasión linfovascular o necrosis deben ser clasificados como sarcoma pleomórfico dérmico»36. Por otro lado, y aunque es una situación infrecuente, están descritos casos de FXA sin invasión subcutánea que hacen recidivas con invasión de estructuras más profundas3 o incluso metástasis8, de modo que, aunque es una situación infrecuente, no es imposible.
Por último, actualmente se considera que el diagnóstico de FXA debe ser realizado con base en una pieza de resección completa y no con biopsias parciales, debido a que es muy importante valorar el patrón de infiltración, la invasión de estructuras y el resto de criterios de SPD1. Aunque se ha tratado de recoger de forma sistematizada, el único estudio que especifica el número de casos diagnosticados con borde profundo afectado es el de Iglesias-Pena et al. (2020)3, donde además se describen dos casos de regresión espontánea tras una biopsia incisional. Esto es relevante porque el diagnóstico de FXA y/o SPD, cuando el borde profundo es positivo, entraña siempre cierto grado de incertidumbre. Es necesario establecer unos criterios diagnósticos claros para poder clasificar tumores sugestivos de FXA que regresan tras una biopsia o tumores incorrectamente resecados, con bordes positivos que no permiten una distinción clara entre FXA y SPD, especialmente si la ampliación de márgenes posterior es negativa.
ConclusionesLas series de FXA y SPD publicadas en los últimos años distinguen estos tumores mayoritariamente con base en los criterios histopatológicos de infiltración subcutánea, necrosis tumoral, infiltración perineural e invasión linfovascular. Las series de FXA que especifican criterios histopatológicos estrictos muestran que se trata de una neoplasia de comportamiento benigno, pero carecemos de series largas de pacientes con SPD que nos permitan establecer de forma clara qué características se asocian con recidiva local o metástasis. Los datos publicados hasta el momento no permiten realizar una diferenciación clara entre estos dos tumores a otros niveles, lo que ha hecho que haya autores que los consideren una misma entidad que puede evolucionar de forma agresiva con base en ciertas características de mal pronóstico. Son necesarios nuevos estudios con series largas de pacientes con descripción minuciosa de sus características histopatológicas, los que nos permitan establecer de forma más rigurosa factores de mal pronóstico y así ayudarnos a elegir los tratamientos más adecuados para nuestros pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


